WSC 2022-2023
Conference 13
Case 3
Signalment:
The canine puppies are a mixture of German shepherd, Alaskan malamute, and Norwegian elkhound. It is unknown how large the litters were, but all puppies had clinically healthy littermates. The breeder also reports that a single puppy from a previous litter (also with the same parentage), died at four weeks old.
History:
Three female mixed-breed canine puppies from two sequential litters with the same parents, died at home at less than 5 weeks of age. All animals had a history of poor weight gain, and pale grey, waxy, odorless feces. Puppy #1 (from litter “A”) was approximately 3 weeks old and weighed 555 g. Puppy #2 and #3 (both from litter “B”) were both approximately 4 weeks old and weighed 1235 g and 2000 g, respectively.
Gross Pathology:
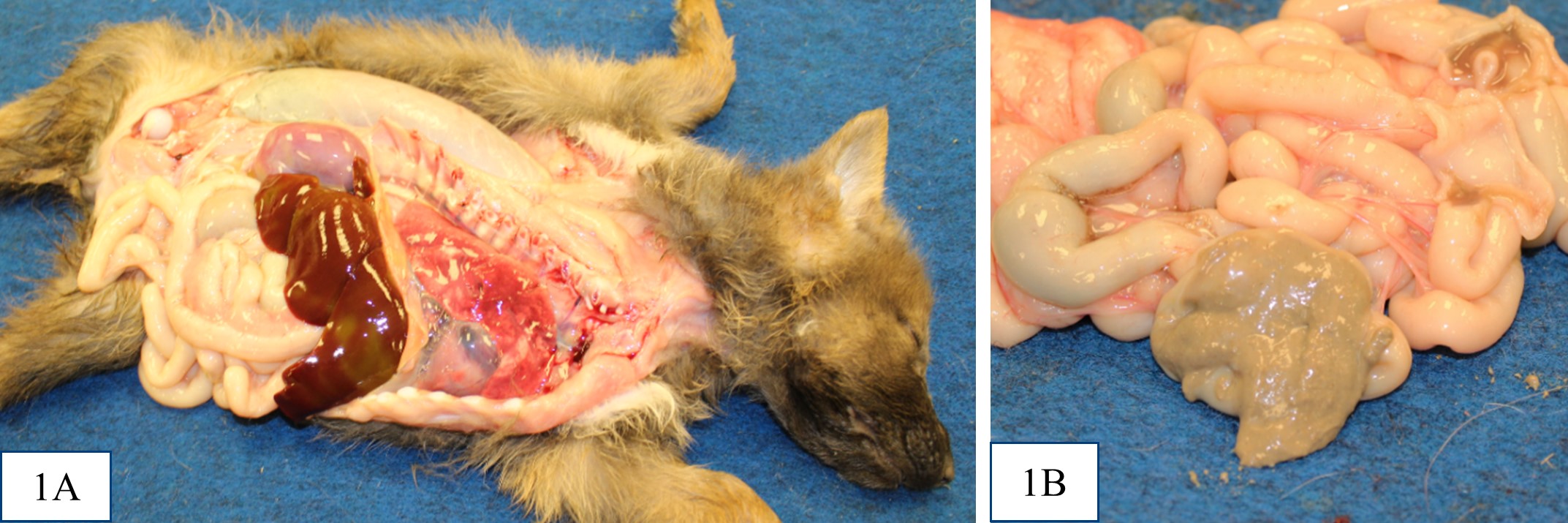
All puppies were in thin body condition, with reduced internal fat stores. The distal small intestine and colon contained thick, pale grey-tan digesta (interpreted as acholic). Puppies #2 and #3 were also noted to be mildly
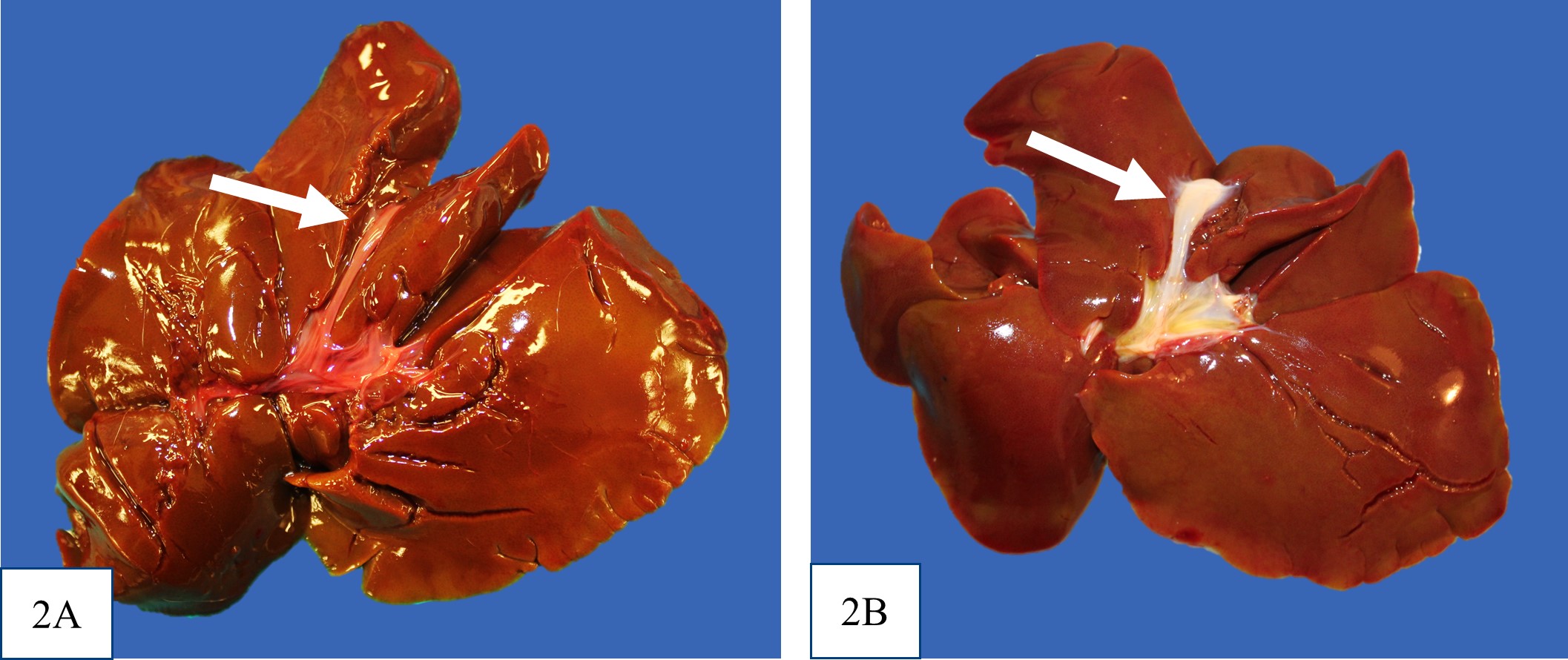
icteric. There was no evidence of extrahepatic portal shunt vessels, and the external anatomy of the livers was normal. In all puppies, at the location of the expected gall bladder and common bile duct, there was a firm tubular structure with no bile content on cut section. It was not recorded in the gross post mortem reports for puppy #1 or #2 whether the major duodenal papilla was present, but the opening of the major duodenal papilla was not grossly appreciable in puppy #3. Puppy #1 had dark black mucous over the rugal folds of the stomach, and similar dark tarry content in some areas of the small intestine and within the colon.
Laboratory Results:
No findings reported.
Microscopic Description:
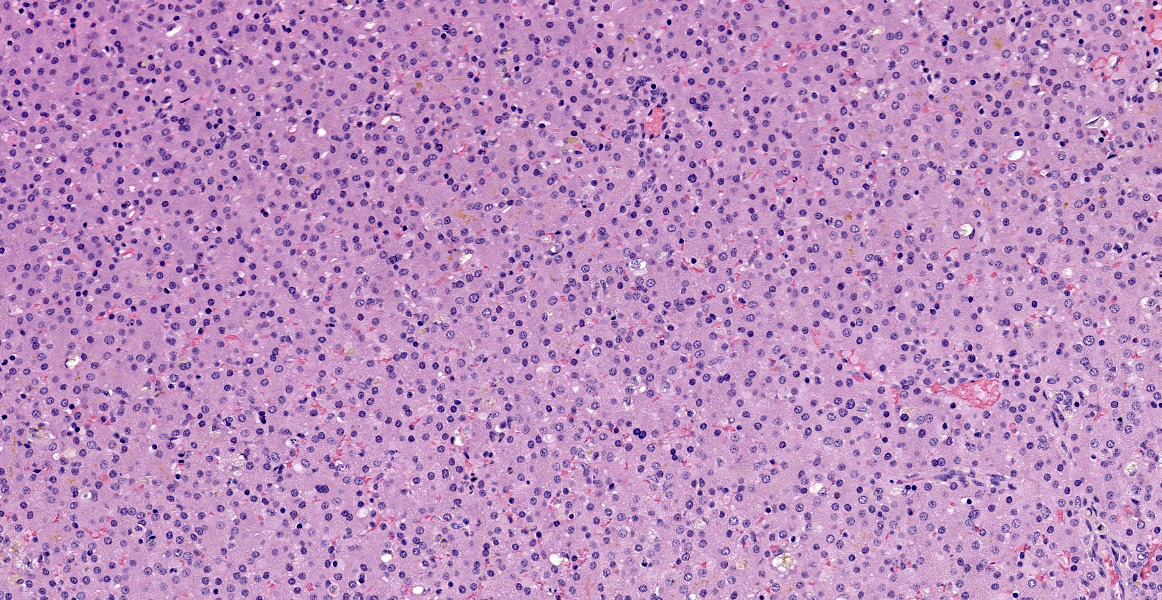
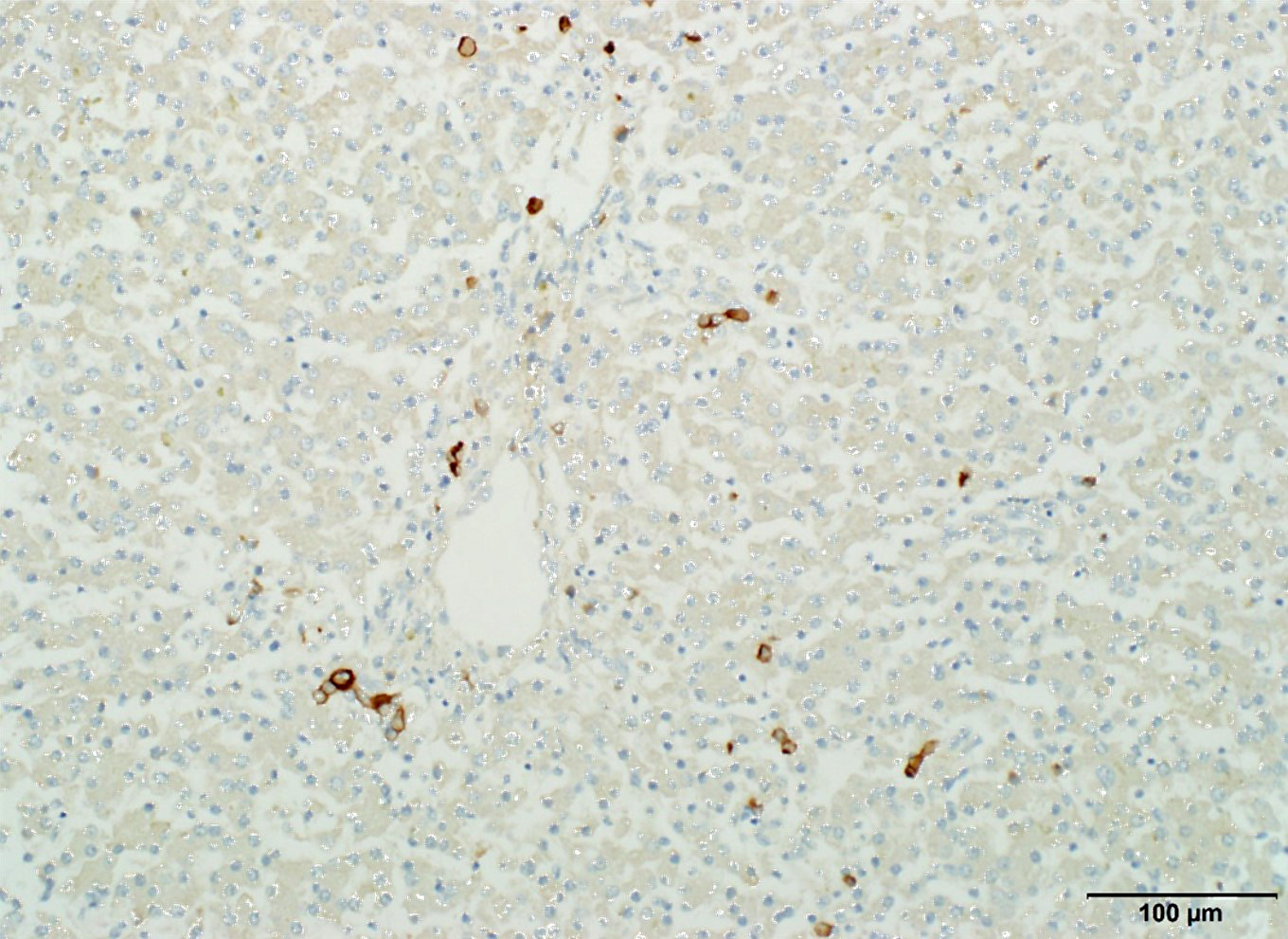
There is abnormal liver architecture, with no appreciable portal triads, as no biliary epithelium was evident. Portal veins were irregularly spaced, and frequently accompanied by small, reduplicated arterioles, often oriented along the limiting plate and occasionally branching out within the hepatic parenchyma. Hepatocytes were arranged in disorganized cords up to three hepatocytes thick, and occasionally in a more sheet-like formation. Where present, sinusoids were often dilated or ectatic, and adjacent hepatocytes were thin and atrophic. There were innumerable bile plugs in canaliculi throughout the parenchyma. Frequently, Kupffer cells and up to 30% of hepatocytes contained scant fine brown granular pigment.
Contributor’s Morphologic Diagnoses:
Congenital atresia of the biliary tract and intrahepatic cholestasis.
Contributor’s Comment:
The three puppies had absence of the gall bladder and intrahepatic bile ducts, with intrahepatic cholestasis. The lack of CK7 positive cells forming distinct tubules throughout the liver confirms the diagnosis of complete absence of intrahepatic bile ducts.
In broad categories, the absence of intrahepatic bile ducts and a gall bladder could be because they never formed or that they had partially or completely formed, and then were later lost. During organogenesis of the liver and biliary system, a part of the endoderm forms the hepatic diverticulum, which subdivides into a cranial portion (pars hepatica) and a caudal portion (pars cystica).4 The gall bladder develops from the pars cystica, and the timing of this development and the precise pathways involved have not been well characterized.15,23 In contrast, the common bile duct, intrahepatic biliary system, and hepatocytes differentiate from bipotential progenitor hepatoblast cells that differentiate in the pars hepatica– the hepatoblasts within the liver parenchyma become hepatocytes, and those at the interface between the parenchyma and the portal mesenchyme surrounding portal veins strongly express biliary-specific cytokeratins and become biliary epithelial cells that form a single layer referred to as the ductal plate.3 In the development of intrahepatic bile ducts, just after day 16.5 of embryogenesis in mice when the ductal plate becomes partially bilayered, small focal dilations appear between the two cells layers, giving rise to bile ducts.7 As the puppies in this case series lacked both a gall bladder and intrahepatic biliary structures, the deviation from normal organ development may have happened at the level of the diverticulum before the subdivision into the pars cystica and pars hepatica, or less likely, there may have been issues in both developmental pathways after the division.
There are a variety of conditions characterized by intrahepatic biliary maldevelopment in humans, which in the literature is referred to as paucity of interlobular bile ducts, intrahepatic biliary hypoplasia or aplasia, intrahepatic biliary atresia, ductular paucity, ductopenia, and ductular hypoplasia.1,9 Complete congenital absence of the intrahepatic bile ducts has been reported in ten children, many of whom died in early childhood. Unlike these puppies, none of the human cases were reported to have an abnormal extrahepatic biliary system except for one child that had fibrotic cystic and common bile ducts (not observed in any of the canine cases).12
Extrahepatic biliary atresia has also been reported in humans, and also rarely in domestic species including dogs, lambs, cats, monkeys, calves and horses.8,13,14,18,20,22,25 The pathogenesis suggested in all species involves progressive fibrosis resulting in disconnection of the common bile duct from the intrahepatic biliary tree, occurring anywhere from the duodenum to the porta hepatis.14,16 Extrahepatic biliary atresia in some affected animals was considered naturally occurring, and in others, occurred following toxin exposure (e.g. experimentally induced with fetal trypan blue exposure in piglets, plant toxins in sheep and calves).14,19 In the two reported cases in young dogs, a distended gall bladder was appreciated, with bile duct obstruction. Cholecystoduodenostomy was performed to surgically circumvent the obstruction in both cases, reportedly with a good short-term clinical outcome in one case.25 In the other case, involving a border collie puppy, the dog died 6 weeks after the cholecystoduodenostomy was performed due to biliary and hepatic toxocariasis, and histology of the common bile duct at the site of the atresia demonstrated replaced of the bile duct by solid fibrous tissue.22 Based on the literature, the extrahepatic biliary atresia previously reported in dogs is different from the puppies described in this case series, as there was no evidence of normal gall bladder or common bile duct structures. Additionally, extrahepatic biliary atresia in all species and humans eventually leads to increased connective tissue within portal areas and surrounding proliferating biliary ductules – as we had no intrahepatic biliary structures or increased connective tissue in our cases, the condition affecting these puppies is distinctly different.2
Congenital absence of a gall bladder has also been reported in dogs, most often affecting small breed dogs, including Chihuahuas (10 of 17 reported cases), toy poodles (3 of 17 reported cases) and Jack Russell terriers (1 of 17 reported cases), and most often accompanied by hypoplasia or absence of some hepatic lobes.21 Histologically, all dogs with gall bladder agenesis had similar findings to extrahepatic biliary atresia, with biliary hyperplasia and portal fibrosis, which does not fit with our case series.21
Very little literature exists on gall bladder development, but there have been a few key pathways identified in intrahepatic biliary epithelium development which may be abnormal in these puppies. The most common cause of intrahepatic biliary maldevelopment in humans is Alagille syndrome, an autosomal dominant disorder with variable expressivity which is also characterized by at least three of the five main clinical manifestations, including chronic cholestasis, vertebral arch defects, pulmonary artery hypoplasia or stenosis, posterior embryotoxon, and a particular set of facial features.10 This syndrome does not include absence of a gall bladder, and these puppies lack the other clinical manifestations of the syndrome, but knowledge of this human condition provides some insight into potential genes and signalling pathways that could be abnormal in this case series.1
Genetically, Alagille syndrome has been characterized by a duplication of exon 6 of the JAG1 gene on chromosome 20, which encodes a Notch1-signalling pathway with importance in embryogenesis.26 Notch signalling has been demonstrated to be required for intrahepatic bile duct and ductal plate formation in mice, and loss of Notch signalling in mouse hepatoblast progenitor cells results in reduction of peripheral intrahepatic bile duct branches postnatally.11,16,24,27 Research of Alagille syndrome has also led to the discovery of transcription factor protein hepatocyte nuclear factor (HNF)-6, which appears to regulate the number of cells that are stimulated to transition from a hepatoblast to a biliary epithelial cell.7 It is suspected that the levels of this protein influence and are influenced by the hepatic mesenchyme, which also plays a vital role in the transition to biliary epithelium.23
Lastly, to explore the possibility that these puppies had biliary trees that had partially or completely formed, and then were later lost, drug-induced “vanishing bile duct syndrome” was briefly considered, which is a human condition that develops following toxin-induced damage to bile ducts.9 Over thirty drugs are implicating this syndrome, which in its chronic form is clinically characterized by prolonged jaundice or chronic cholestasis for more than one year following administration of one of these drugs, and is histologically characterized by a bile to portal tract ratio of less than 0.5.8,9 Although these dogs meet the defined criteria for “ductopenia” as described by the literature, these dogs have no known history of drug exposure, and the canine cases in this series lack portal tracts infiltrated by mononuclear cells (which is commonly seen in the human condition).9 Lastly, again, this phenomenon has also not been reported in association with absence of a formed gall bladder.9
In summary, the present case report demonstrates the existence of a novel canine condition characterized by congenital absence of both intrahepatic bile ducts and the gall bladder and extrahepatic biliary tree. According to medical dictionaries, atresia is defined as “abnormal closing or absence of a tube in the body” whereas aplasia is defined as “lack of growth of tissue”.6 Using this nomenclature, arguably both “extrahepatic and intrahepatic biliary atresia” and “extrahepatic and intrahepatic biliary aplasia” could be appropriate when describing these puppies. However, considering that we were unable to unequivocally characterize the CK7 positive duct in the submucosa of the duodenum as being pancreatic or hepatopancreatic in origin, and leaving open the possibility that it may reflect a small portion of an intact common bile duct (which would imply that there is a regionally extensive absence of the extrahepatic biliary tree), “extrahepatic and intrahepatic biliary atresia” may be more appropriate in this case. As multiple litters with the same parentage were affected, we suspect an underlying genetic cause.
In summary, the present case report demonstrates the existence of a novel canine condition characterized by congenital absence of both intrahepatic bile ducts and the gall bladder and extrahepatic biliary tree. According to medical dictionaries, atresia is defined as “abnormal closing or absence of a tube in the body” whereas aplasia is defined as “lack of growth of tissue”26. Using this nomenclature, arguably both “extrahepatic and intrahepatic biliary atresia” and “extrahepatic and intrahepatic biliary aplasia” could be appropriate when describing these puppies. However, considering that we were unable to unequivocally characterize the CK7 positive duct in the submucosa of the duodenum as being pancreatic or hepatopancreatic in origin, and leaving open the possibility that it may reflect a small portion of an intact common bile duct (which would imply that there is a regionally extensive absence of the extrahepatic biliary tree), “extrahepatic and intrahepatic biliary atresia” may be more appropriate in this case. As multiple litters with the same parentage were affected, we suspect an underlying genetic cause.
Contributing Institution:
Ontario Veterinary College, Department of Pathobiology, Guelph
https://ovc.uoguelph.ca/pathobiology/
JPC Diagnosis:
Liver: Biliary aplasia.
JPC Comment:
The contributor provides an excellent review of embryologic development of the liver and gallbladder as well as the existing literature on gall bladder agenesis and biliary atresia. Another novel presentation of gall bladder agenesis was recently reported by Mestrinho et al in a 1-year-old male French bulldog. In addition to gall bladder agenesis, the dog had an umbilicobiliary fistula that resulted in persistent yellow discharge from the umbilicus.17 A duct-like structure connected the umbilicus to the common bile duct; histopathologic examination of the resected structure revealed similar features to a gall bladder or bile duct.17 The authors speculated that the pars cystica of the hepatic diverticulum became trapped during invagination of umbilical cord structures and subsequently formed a cutaneous fistula.17
Dr. Cullen and conference participants discussed the greater amount of bile but lesser degree of hepatocellular damage in this case compared to case 2 (lantana toxicity in a goat). This is possibly because bile canaliculi are intact, while in the lantana toxicity case, bile canaliculi are injured, and liberated bile salts caused damage to hepatocellular membranes. Participants elected to use “aplasia” as a morph due to the complete lack of biliary profiles; most other forms of biliary atresia which frequently feature small CK7 positive biliary profiles.
References:
- Alagille D, Estrada A, Hadchouel M, Gautler M, Odievre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110(2):195-200.
- Bassett MD, Murray KF. Biliary atresia: recent progress. J Clin Gastroenterol. 2008;42(6):720.
- Bastianello SS, Nesbit JW. The pathology of a case of biliary atresia in a foal. J S Afr Vet Assoc. 1986;57(2):117-120.
- Bedi N, Bond-smith G, Kumar S, Hutchins R. Gallbladder agenesis with choledochal cyst—a rare association: a case report and review of possible genetic or embryological links. BMJ Case Rep. 2013;2013:bcr2012006786.
- Bouwens L. Cytokeratins and cell differentiation in the pancreas. J Pathol A J Pathol Soc Gt Britain Irel. 1998;184(3):234-239.
- Collin PH, ed. Dictionary of Medicine. 2nd ed. New York: Routledge, Taylor & Francis Group; 2014.
- Clotman F, Lannoy VJ, Reber M, et al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129(8):1819-1828.
- Degott C, Feldmann G, Larrey D, et al. Drug?induced prolonged cholestasis in adults: a histological semiquantitative study demonstrating progressive ductopenia. Hepatology. 1992;15(2):244-251.
- Desmet VJ. Vanishing bile duct syndrome in drug-induced liver disease. J Hepatol. 1997;26:31-35.
- Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29(3):822-829.
- Geisler F, Nagl F, Mazur PK, et al. Liver?specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology. 2008;48(2):607-616.
- Haas L, Dobbs RH. Congenital absence of the intrahepatic bile ducts. Arch Dis Child. 1958;33(171):396.
- Hampson E, Filippich LJ, Kelly WR, Evans K. Congenital biliary atresia in a cat: a case report. J Small Anim Pract. 1987;28(1):39-48.
- Harper PAW, Plant JW, Ungers DB. Congenital biliary atresia and jaundice in lambs and calves. Aust Vet J. 1990;67(1):18-22.
- Lemaigre F. Development of the biliary tract. Mech Dev. 2003;120(1):81.
- Lozier J, McCright B, Gridley T. Notch signaling regulates bile duct morphogenesis in mice. PLoS One. 2008;3(3):e1851.
- Mestrinho LA, Monteiro C, Sobral C, Travancinha J, Niza MM. A case of a congenital umbilicobiliary fistula associated with gallbladder agenesis in a dog. Rev Bras Med Vet. 2022; 44:1-5.
- Rosenberg DP, Morecki R, Lollini LO, Glaser J, Cornelius CE. Extrahepatic biliary atresia in a rhesus monkey (Macaca mulatto). Hepatology. 1983;3(4):577-580.
- Rosenkrantz JG, Lynch FP, Frost WW. Congenital anomalies in the pig: Teratogenic effects of trypan blue. J Pediatr Surg. 1970;5(2):232-237.
- Ruíz-Ramírez JA, García-Márquez LJ, Bedolla-Alva MA, et al. Congenital biliary atresia in a Beefmaster calf. Brazilian J Vet Pathol. 2016;9(3):93-97.
- Sato K, Sakai M, Hayakawa S, et al. Gallbladder Agenesis in 17 Dogs: 2006–2016. J Vet Intern Med. 2018;32(1):188-194.
- Schulze C, Rothuizen J, Sluijs FJ van, Hazewinkel HAW, Van Den Ingh T. Extrahepatic biliary atresia in a border collie. J Small Anim Pract. 2000;41(1):27-30.
- Shiojiri N. Development and differentiation of bile ducts in the mammalian liver. Microsc Res Tech. 1997;39(4):328-335.
- Sparks EE, Perrien DS, Huppert KA, Peterson TE, Huppert SS. Defects in hepatic Notch signaling result in disruption of the communicating intrahepatic bile duct network in mice. Dis Model Mech. 2011;4(3):359-367.
- Thiel C, Steinbach S, Schmidt M, et al. Extrahepatic biliary atresia in a 4?week?old pug. Vet Surg. 2015;44(1):35-40.
- Uberos J, Moreno L, Muñoz-Hoyos A. Hypertension and Biliary Ductopenia in a patient with Duplication of exon 6 of the JAG1 Gene. Clin Med Insights Pediatr. 2012;6:CMPed-S9621.
- Zong Y, Panikkar A, Xu J, et al. Notch signaling controls liver development by regulating biliary differentiation. Development. 2009;136(10):1727-1739.