Signalment:
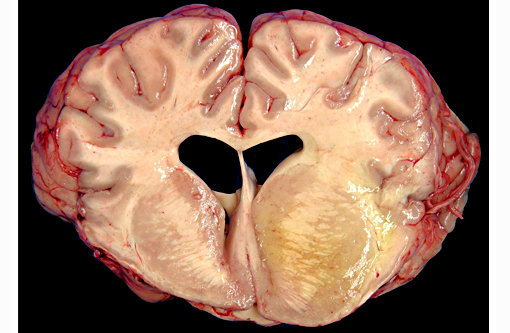
Gross Description:
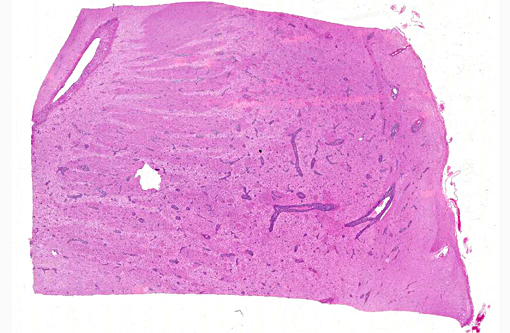
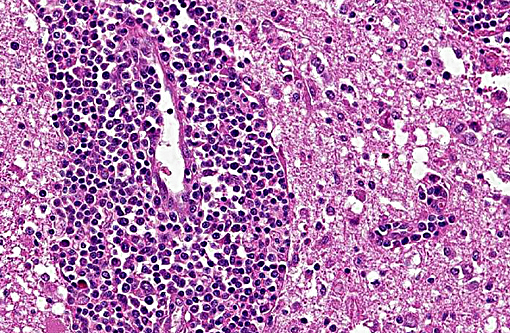
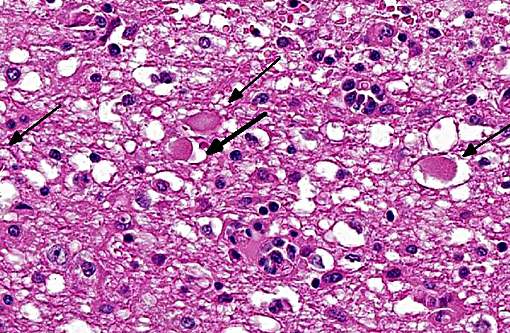
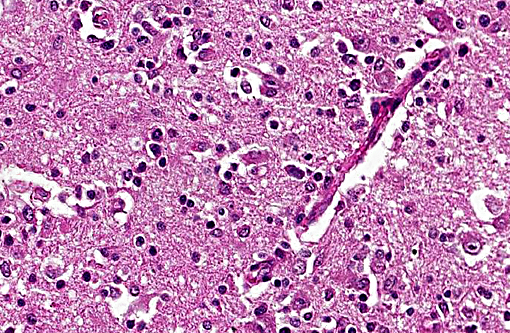
Histopathologic Description:
The brain lesions affected mainly the white matter and were characterized by moderate to severe perivascular lymphoplasmacytic menin-goencephalitis, edema and necrosis. Lympho-cytes and plasma cells, often with intracy-toplasmic Russell bodies (Mott cells), greatly expanded the Virchow-Robin spaces and extended into the surrounding neuropil. Lesions in the spinal cord tended to wane from cranial to caudal. Organisms of T. evansi were not detected in the HE stained sections of brain, but numerous T. evansi organisms or fragments of these organisms were detected in formalin-fixed, paraffin-embedded sections of the brain by immunohistochemistry through anavidin-biotin-peroxidase complex immunoperoxidase method using rabbit anti-T. evansi, diluted 1:1000 as the primary antibody (the immunohistochemistry tests were performed at the Prairie Diagnostic Service, University of Saskatchewan, Western College of Veterinary Medicine). The parasites were observed in the perivascular spaces an in the neuropil.
Morphologic Diagnosis:
Etiologic diagnosis: Protozoal encephalitis
Etiology: Trypanosoma evansi
Name of the condition: Trypanosomiasis
Lab Results:
Condition:
Contributor Comment:
Trypanosomiasis in horses is characterized by intermittent fever, anemia, progressive weakness, loss of body condition, and unstable gait.(21) Neurologic signs have been described occasionally in horses in the terminal phase of natural infection by T. evansi.(21) Trypanosomiasis by T. evansi is enzootic in horses from the Midwestern Brazil, mainly the Pantanal region.(3,4,6,9,22, 23)
T. evansi induces a wasting disease with a protracted clinical course(28) associated with anemia and instability of pelvic limbs in horses, camels, and dogs.(4) Natural infection by T. evansi rarely is recognized as a cause of encephalitis in horses.(21) Neurologic signs have been reported in cattle(27) and hog deer(26) naturally infected by T. evansi; however, the histopathologic changes in the brain of these species were not fully described. Mild lymphoplasmacytic meningo-encephalitis was reported in horses, donkeys, dogs, goats,(2) coatis (Nasua nasua),(9) and buffalo(25) experimentally infected by T. evansi. The horse of this report and the other necropsied horses in the current outbreak had a severe lympho-plasmacytic meningoencephalitis with marked edema and variable necrosis. The lesions in the brain of these horses resembled those described in horses infected by Trypanosoma brucei brucei,(14,17) which causes a disease known as nagana, and those described for human trypano-somiasis caused by Trypanosoma brucei gambiense and Trypanosoma brucei rhodesiense. (13,16)
T. evansi antigen was detected by immunohistochemistry in the brain of 8 of 9 horses affected in the outbreak, including the brain of the horse of this report. The protozoa were detected within blood vessels, in perivascular spaces, and in the brain parenchyma. This suggests that trypanosomes invaded the brain parenchyma and were responsible for the lesions observed here. The pathogenesis of cerebral necrosis was not elucidated since thrombosis was absent. Neither the mechanism by which trypanosomes entered the neuroparenchyma was determined. The introduction of T. evansi into a na+â-»ve population, as was the case here, could explain the development of fatal trypanosomiasis with severe encephalitis. The reason for the sudden appearance of the disease in this region of Brazil, where it was previously unreported, is unknown, but it could have been introduced by transportation of infected horses or migration of capybaras from regions where trypanosomiasis is enzootic in this country. A factor that may have contributed to the development of severe encephalitis was that these affected horses were treated with subtherapeutic doses of diminazene aceturate and other antitrypanosomal drugs.(19,20) Several studies have demonstrated that the use of subtherapeutic doses of diminazene aceturate may prolong survival of horses experimentally infected by T. brucei spp. however this faulty procedure results in subsequent invasion of the central nervous system by the organisms which induce necrotizing encephalitis.(8,10,11,15,18,24) Diminazene aceturate clears trypanosomes from tissues except those localized on the central nervous system,(19) because the drug does not cross the blood-brain barrier(11,17) allowing trypanosomes harbored in the central nervous system to survive antitrypanosomal therapy; a change in these organisms surface glycoproteins favor recurrent parasitemias by variants of the organism.(20)
How trypanosomes penetrate the blood-brain barrier is not clear, but several mechanisms have been proposed: (i) entrance through sites where the blood-brain barrier is incomplete, such as sensory ganglia and circumventricular organs;(16), (ii) increase in vascular permeability due deposition of immune complexes in the choroid plexus; and (iii) opening of intercellular tight junctions of the ependymal lining of the ventricular system by toxins released by the parasite.(12) It is believed that invasion of the central nervous system by trypanosomes occurs where the blood-brain barrier has been disrupted, either directly by the parasites or by release of chemical mediators, such as cytokines and proteases.(7,13,15) An interaction between the trypanosomes and the host response could promote tissue damage and facilitate the entry of parasites into the central nervous system.(13) This scenario is supported by the histopathologic changes and immunohistochemistry observe in the horse of this report. Trypanosomiasis due to T. evansi should be considered in the differential diagnosis of encephalitis in horses in regions where the disease is enzootic.
T. evansi should be considered in the differential diagnosis of encephalitis in horses in regions where the disease is enzootic. The list of differentials should include equine herpesvirus type 1 myeloencephalopathy, Eastern equine encephalitis, Western equine encephalitis, Venezuelan equine encephalitis, equine protozoal myeloencephalitis, West Nile virus infection, and rabies. The nature and distribution of lesions in the central nervous system of horses with naturally occurring T. evansi infection should help to distinguish trypanosomiasis from viral or other protozoal infections.
JPC Diagnosis:
Conference Comment:
T. evansi is the causative agent of surra, which originated in Africa, but is also present across the Middle East, Asia, in Central and South America and has also been reported in Russia. It is thought to derive from genetic modification (a deletion) in T. brucei. It has a wide host range, indeed the widest among salvarian trypanosomes, including camels, dogs, horses, deer, llamas, cattle, sheep, goats, cats, pigs, and elephants among others. Disease is particularly devastating in camels and horses, often proving fatal in the absence of treatment. Clinical signs can vary dramatically between species and even between individuals of the same species. Infection in camels can occur acutely as in horses, but often has a more protracted course with signs such as intermittent fever, weakness, anorexia, weight loss, abortion, anemia, edema and petechial and ecchymotic hemorrhages. All age groups are affected and the urine of infected camels apparently has a specific odor, which may allow diagnosis of the disease. Dogs are also highly susceptible to infection and death may result at 1-4 weeks post infection.
Trypanosomes have developed mechanisms which allow them to both evade the immune system and cause immunosuppression. This includes antigenic variation in main membrane surface glycoproteins, variant surface glycoprotein (VSG), which forces the host to constantly redevelop their humoral response. The immunosuppressive effects of T. evansi infection are not fully understood but may involve modulation of macrophage activity, decreased responsiveness of lymphocytes, and/or changes in the CD4:CD8 lymphocyte ratio; they may even be capable of eliminating memory B cells. The complex immunosuppressive or immunomodulatory mechanisms of T. evansi have created significant barriers in developing effective vaccines and maintaining effective treatments.(5)
References:
1. Camargo RE, Uzcanga GL, Bubis J. Isolation of two antigens from Trypanosoma evansi that are partially responsible for its cross-reactivity with Trypanosoma vivax. Vet Parasitol. 2004;123:67-81.
2. Dargantes AP, Campbell RSF, Copeman DB, et al. Experimental Trypanosoma evansi infection in the goat. II. Pathology. J Comp Pathol. 2005;133:267-276.
3. Davila AMR, Silva RAMS. Animal trypanosomiasis in South America. Current status, partnership, and information technology. Ann N Y AcadSci. 2000;916: 199-212.
4. Davila AMR, Souza SS, Campos C, Silva RA. The seroprevalence of equine trypanosomiasis in the Pantanal. Mem Inst Oswaldo Cruz 1999;94:199-202.
5. Desquesnes M, Holzmuller P, Lai DH, Dargantes A, Lun ZR, Jittaplapong S. Trypanosoma evansi and surra: a review and perspectives on origin, history, distribution, taxonomy, morphology, hosts, and pathogenic effects. Biomed Res Int. 2013;194176:1-22.
6. Franke CR, Greiner M, Mehlitz D. Investigations on naturally occurring Trypanosoma evansi infections in horses, cattle, dogs and capybaras (Hydrochaeris hydrochaeris) in Pantanal de Pocon+â-¬ (Mato Grosso, Brazil). Acta Trop. 1994;58:159-69.
7. Girard M, Bisser S, Courtioux B, Vermot-Desraches C, Bouteille B, Wijdenes J, Preudhomme JL, Jauberteau MO. In vitro induction of microglial and endothelial cell apoptosis by cerebrospinal fluids from patients with human African trypanosomiasis. Int J Parasitol. 2003;33:713720.
8. Grab DJ, Nikolskaia O, Kim YV, Londsdale-Eccles JD, Ito S, Hara T, Fukuma T, Nyarko E, Kim KJ, Stins MF, Delannoy MJ, Rodgers J, Kim KS. African trypanosome interactions with an in vitro model of the human blood-brain barrier. J Parasitol. 2004;90:970-979.
9. Herrera HM, D+â-ívila AMR, Norek A, Abreu UG, Souza SS, DAndrea PS, Jansen AM. Enzootiology of Trypanosoma evansi in Pantanal, Brazil. Vet Parasitol. 2004;125:263-275.
10. Keita M, Bouteille B, Enanga B, et al. Trypanosoma brucei brucei: a long-term model of human African trypanosomiasis in mice, meningoencephalitis, astrocytosis, and neurologic disorders. Exp Parasitol. 1997;85:183-192.
11. Kennedy PGE, Rodgers J, Jennings FW, et al. A substance P antagonist, RP-67, 580, ameliorates a mouse meningoencephalitic response to Trypanosoma brucei brucei. Proc Natl Acad Sci USA 1997;94:4167-4170.
12. Lambert PH, Berney M, Kazyumba G. Immune complexes in serum and in cerebrospinal fluid in African trypanosomisis. Correlation with polyclonal B cell activation and with intracerebral immunoglobulin synthesis. J Clin Invest. 1981;67:77-85.
13. Londsdale-Eccles JD, Grab DJ. Trypanosome hydrolases and the blood-brain barrier. Trends Parasitol 2002;18:17-19.
14. Losos GJ, Ikede BD. Review of pathology of diseases of domestic and laboratory animals caused by Trypanosomacongolensis, T. vivax, T. brucei, T. rhodesiense and T. gambiense. Vet Pathol 1972;9(Suppl):1-71.
15. Masocha W, Robertson B, Rottenberg ME, et al. Cerebral vessel laminins and IFN-c define Trypanosoma brucei brucei penetration of blood-brain barrier. J Clin Invest. 2004;114:689-694.
16. Mhlanga JDM, Bentivoglio M, Kristensson K. Neurobiology of cerebral malaria and African sleeping sickness. Brain Res Bull 1997;44:579-589.
17. Moulton JE. Relapse after chemotherapy in goats experimentally infected with Trypanosoma brucei: pathological changes in central nervous system. Vet Pathol. 1986;23:21-28.
18. Ouwe-Missi-Oukem-Boyer O, Mezui-Me-Ndong J, BodaC, et al. The vervet monkey (Chlorocebusa ethiops) as an experimental model for Trypanosoma brucei gambiense human African trypanosomiasis: a clinical, biological and pathological study. Trans R Soc Trop Med Hyg. 2005;100:427-436.
19. Rodrigues A, Fighera RA, Souza TM, et al. Surtos de tripanossom+â-¡ase em eq+â-+inos no Rio Grande do Sul: aspectos epidemiol³gicos, cl+â-¡nicos, hematol³gicos e patol³gicos. Pesq Vet Bras. 2005;25:239-249.
20. Rodrigues A, Fighera R. A., Souza TM, et al. Neuropathology of naturally occurring Trypanosoma evansi infection of horses. Vet Pathol. 2009;46:251-258.
21. Seiler RJ, Omar S, Jackson AR. Meningoencephalitis in naturally occurring Trypanosoma evansi infection (surra) of horses. Vet Pathol. 1981;18:120-122.
22. Silva RAMS, Herrera HM, Domingos LBS, et al. Pathogenesis of Trypanosoma evansi infection in dogs and horses: hematological and clinical aspects. Ci+â-¬ncia Rural 1995;25:233-238.
23. Silva RAMS, Seidl A, Ramirez L, D+â-ívila AMR.Trypanosoma evansi e Trypanosoma vivax: Biologia, Diagn³stico e Controle. Embrapa Pantanal, Corumb+â-í, Brazil, 2002.
24. Sternberg JM, Rodgers J, Bradley B, Maclean L, Murray M, Kennedy P. Meningoencephalitic African trypanosomiasis: brain IL-10 and IL-6 are associated with protection from neuro-inflammatory pathology. J Immunol. 2005;167:81-89.
25. Sudarto MW, Tabel H, Haines DM. Immunohistochemical demonstration of Trypanosoma evansi in tissues of experimentally infected rats and a naturally infected water buffalo (Bubalus bubalis). J Parasitol. 1990;76:162-167.
26. Tuntasuvan D, Mimapan S, Sarataphan N, et al. Detection of Trypanosoma evansi in brains of the naturally infected hog deer by streptavidine-biotin immunohistochemistry. Vet Parasitol. 2000;87:223-230.
27. Tuntasuvan D, Sarataphan N, Nishikawa H. Cerebral trypanosomiasis in native cattle. Vet Parasitol. 1997;73:357-363.
28. Ventura RM, Takata CSA, Silva RAMS, et al.Molecular and morphological studies of Brazilian Trypanosoma evansi stocks: the total absence of kDNA in trypanosomes from both laboratory stocks and naturally infected domestic and wild mammals. J Parasitol. 2000;86:1289-1298.