Signalment:
Gross Description:
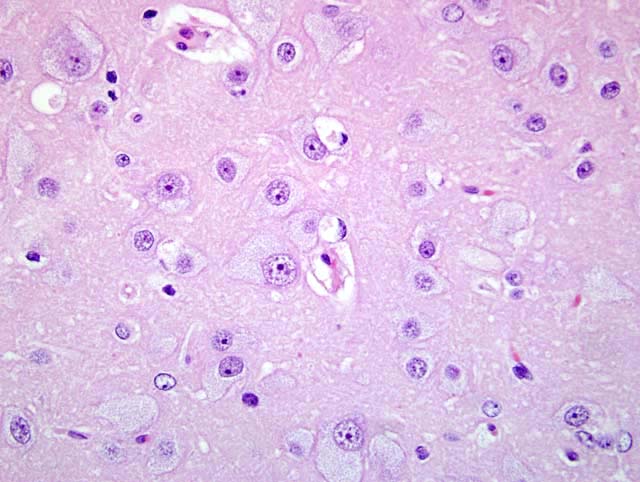
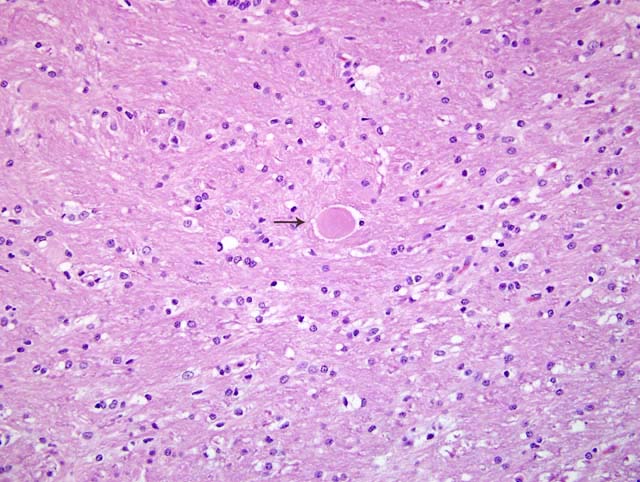
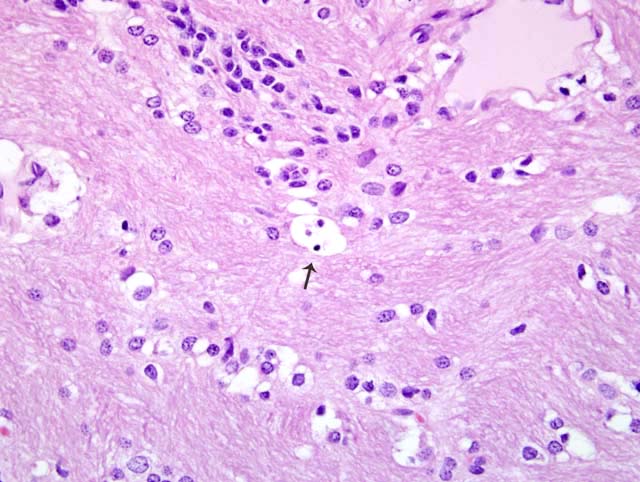
Histopathologic Description:
Morphologic Diagnosis:
Lab Results:
Condition:
Contributor Comment:
Biochemically, the inherited lysosomal storage diseases can be broadly divided into sphingolipidoses, cholesterol ester storage disease, glycoproteinoses, glycogenoses, mucopolysaccharidoses, mucolipidoses and ceroid-lipofuscinoses.3 Sphingolipidoses comprise diseases in which there is a failure to properly catabolize various complex lipids derived from ceramide.3 These include GM1 and GM2 gangliosidosis, galactocerebrosidosis, glucocerebrosidosis, sphingomyelin lipidosis and galactosialidosis.3 GM2 ganglioside is a cell membrane glycolipid which is catabolized by the action of N-acetylyhexosaminidase and GM2 activator (GM2A) protein.6,7 N-acetylyhexosaminidase exists as a dimer in two forms, __ (hexosaminidase A) and __ (hexosaminidase B). Improper functioning of either one of these subunits (_ or _) or GM2A results in accumulation of GM2 ganglioside in the lysosomes of neurons leading to a progressive deterioration of the central nervous system (GM2 gangliosidosis).6,7 Tay-Sachs disease of humans is a GM2 gangliosidosis caused by a mutation in the _ subunit resulting in the deficiency of hexosaminidase A, whereas Sandhoff disease is caused by mutation of _ subunit which affects both hexosaminidase A and B.3 GM2 gangliosidosis has been previously described in German shorthaired pointer and Japanese Spaniel dogs, domestic short haired and Korat cats, and Muntjak deer.1,3
GM1 gangliosidosis (generalized gangliosidosis) is caused by the deficiency of _ galactosidase which cleaves the terminal galactose from GM1 ganglioside. GM1 gangliosidosis has been described in dogs, cats, cattle and sheep.3 Galactocerebrosidosis (globoid cell leukodystrophy, Krabbe disease of humans) is primarily a leukodystrophy and has been described in dogs, cats and sheep. Galactocerebrosides are found in myelin sheaths and the deficiency of galactocerebrosidase causes accumulation of the lipids in the macrophage-like -�-�globoid cells and in oligodendrogial cells.3 Glucocerebrosidosis (Gaucher disease of humans) is caused by a deficiency in the lysosomal enzyme glucocerebrosidase, a _-glucosidase, which results in the accumulation of the lipid glucocerebroside in neurons and in macrophages. The disease has been described in Sydney Silky dogs.3 Sphingomyelin lipidosis (Niemann-Pick disease of humans) is caused by a deficiency of sphingomyelin phosphodiesterase (sphingomyelinase) which results in the accumulation of sphingomyelin in neurons and macrophages. The disease has been described in cats and a single case in miniature Poodle dog.3
JPC Diagnosis:
Conference Comment:
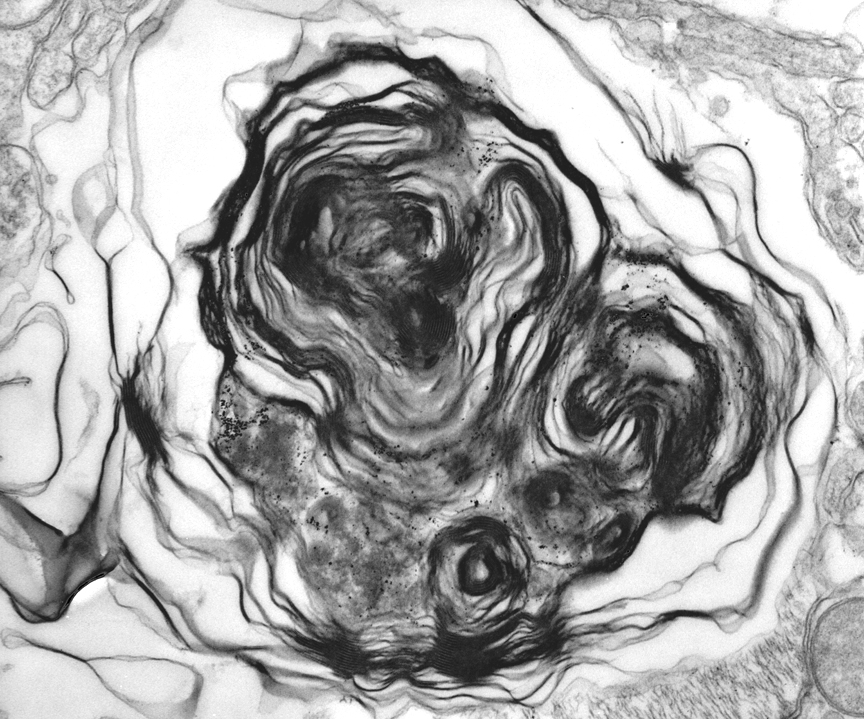
Neurons of the central and autonomic nervous system and retina are primarily affected.5 Typical histopathologic findings in GM2 gangliosidosis include swollen neurons with cytoplasmic vacuoles consisting of lysosomes distended with accumulated gangliosides. Oil red O and Sudan black B may stain storage material in astrocytes and macrophages.3,5 With electron microscopy (EM), the cytoplasmic inclusions are visualized and consist of membrane layers forming "onion-skin" whorls within the lysosomes.5
Lysosomal storage bodies visualized on EM are not considered characteristic of a particular storage disorder. In general, GM1 and GM2 may form similar concentric lamellations, whereas in mucopolysaccharide storage diseases, membranous stacks (zebra bodies) are more common.3 Abnormal twisted tubular structures are often seen in glucocerebrosidosis and galactocerebrosidosis, while glycogen particles may be seen in the glycogen storage diseases.3
References:
2. Hasegawa D, Yamato O, Kobayashi M, Fujita M, Nakamura S, Takahashi K, Satoh H, Shoda T, Hayashi D, Yamasaki M, Maede Y, Arai T, Orima H: Clinical and molecular analysis of GM2 gangliosidosis in two apparent littermate kittens of the Japanese domestic cat. J Feline Med Surg 9:232-237, 2007
3. Jolly RD, Walkley SU. Lysosomal storage diseases of animals: An Essay in Comparative Pathology. Vet Pathol 34:527-548, 1997
4. Kroll RA, Pagel MA, Roman-Goldstein S, Barkovich J, D'Agostino AN, Neuwelt EA: White matter changes associated with feline GM2 gangliosidosis (Sandhoff disease): Correlation of MR findings with pathologic and ultrastructural abnormalities. Am J Neuroradiol 16:1219-1226, 1995
5. Kumar V, Abbas, AK, Fausto N: Genetic disorders. In: Robbins and Cotran Pathologic Basis of Disease, 7th ed., pp. 158-168. Elsevier Saunders, Philadelphia, PA, 2005
6. Martin DR, Krum BK, Varadarajan GS, Hathcock TL, Smith BF, Baker HJ. An inversion of 25 base pairs causes feline GM2 gangliosidosis variant. Exp Neurol. 187:30-37, 2004
7. Martin DR, Cox NR, Morrison NE, Kennamer DM, Peck SL, Dodson AN, Gentry AS, Griffin B, Rolsma MD, Baker HJ. Mutation of the GM2 activator protein in a feline model of GM2 gangliosidosis. Acta Neuropathol 110:443-50 2005
8. Yamato O, Satoh H, Matsuki N, Ono K, Yamasaki M, Maede Y: Laboratory diagnosis of canine GM2-gangliosidosis using blood and cerebrospinal fluid. J Vet Diagn Invest 16:39-44, 2004
9. Yamato O, Matsunaga S, Takata K, Uetsuka K, Satoh H, Shoda T, Baba Y, Yasoshima A, Kato K, Takahashi K, Yamasaki M, Nakayama H, Doi K, Maede Y, Ogawa H: GM2-gangliosidosis variant O (Sandhoff-like disease) in a family of Japanese domestic cats. Vet Rec 155:739-744, 2004