Joint Pathology Center
Veterinary Pathology Services
Wednesday Slide Conference
2017-2018
Conference 5
September 27th, 2017
CASE IV: P16-555 (JPC 4101307).
Signalment: Juvenile, male, domestic pig, Sus scrofa domesticus, porcine.
History: This case is one out of four pigs from an unvaccinated control group of a vaccine development study. The pigs were experimentally infected i.m. with 106 TCID50 of classical swine fever virus wild type strain Alfort/Tuebingen. They were humanely killed after developing clinical disease 15 days post infection including fever up to 41°C, apathy, changed defecation and dermal macula.
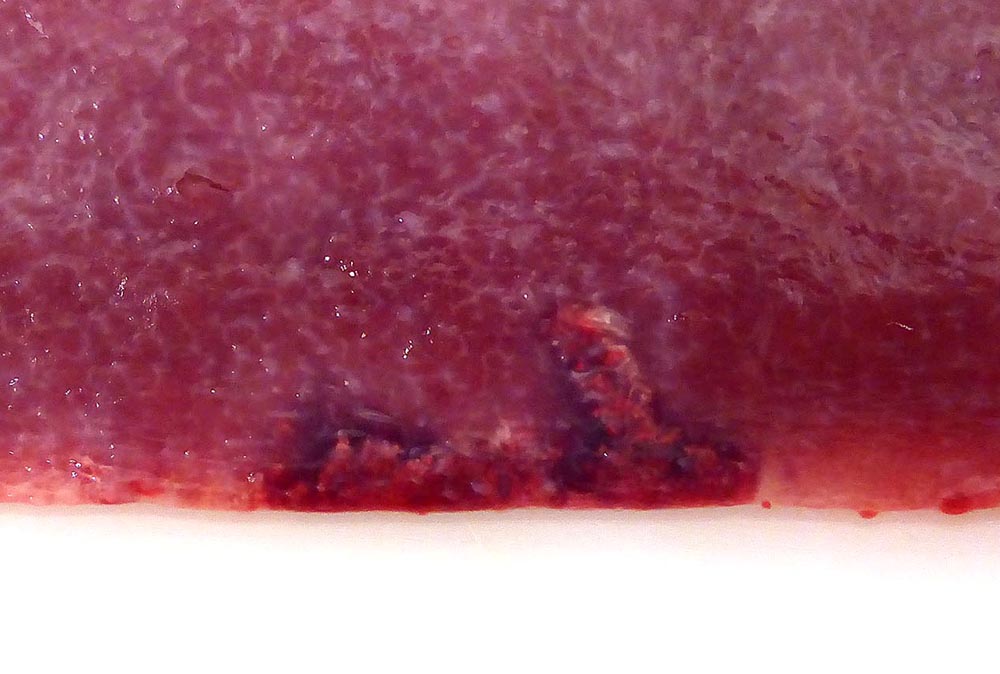
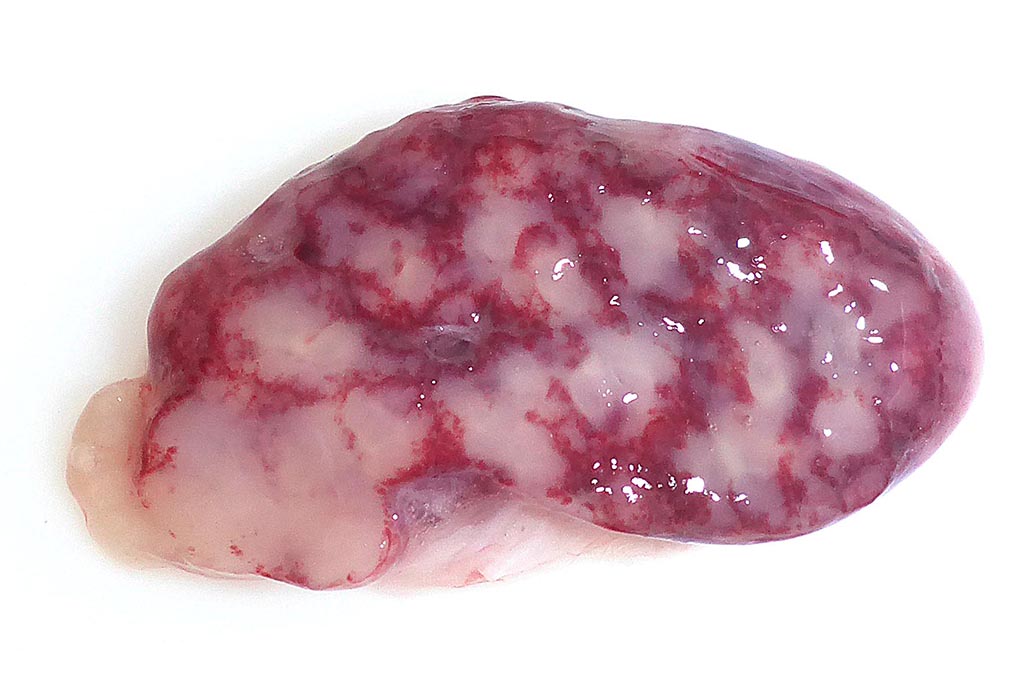
Gross Pathology: The splenic surface showed multifocal, distinct, flame-shaped white streaks with a variably distinct peripheral plum-colored border extending 2-3 mm from and perpendicular to the acute angle. Cut sections revealed roughly wedge-shaped white areas with a variably distinct peripheral plum-colored border at the acute angle, interpreted as mild, multifocal, (ischemic) infarcts.
Other macroscopic changes in this pig were mild hydrothorax and ascites, mild, multifocal, pleural fibrosis with synechiae between parietal and visceral pleura, and moderate hyperplasia of the lymphocentrum bronchiale.
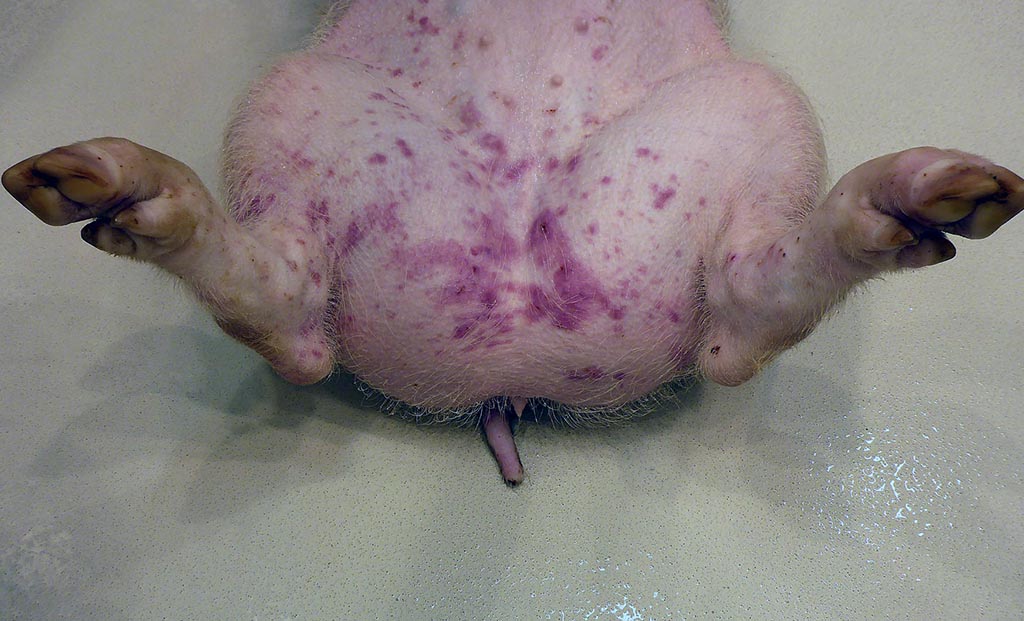
Macroscopic changes in the other pigs included mild to moderate ascites which was the only constant change observed in all four animals. Further important macroscopic changes included moderate, multifocal, irregular cherry red dermal maculae in the inguinal area in two pigs, and peripherally cherry red and white marbled Lymphonodi hepatici et gastrici, interpreted as blood-filled sinusoids in one pig.
Laboratory results: Classical swine fever genomic RNA was detected in multiple samples of this pig beginning 5 days post infection and up to day 15 using diagnostic real-time RT-PCR.5 The results were clearly positive with threshold cycle values of 25.12 on the day of killing.
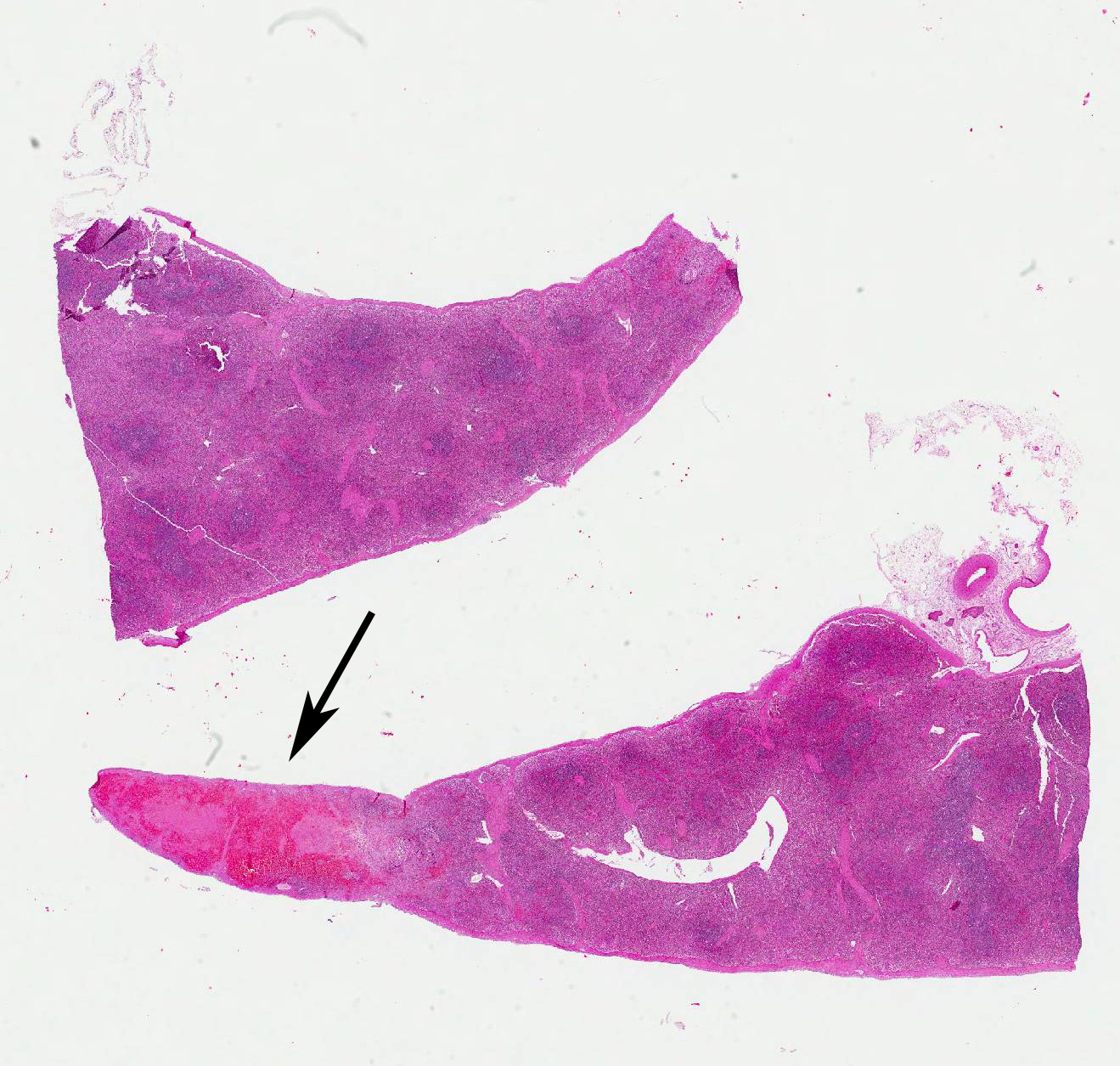
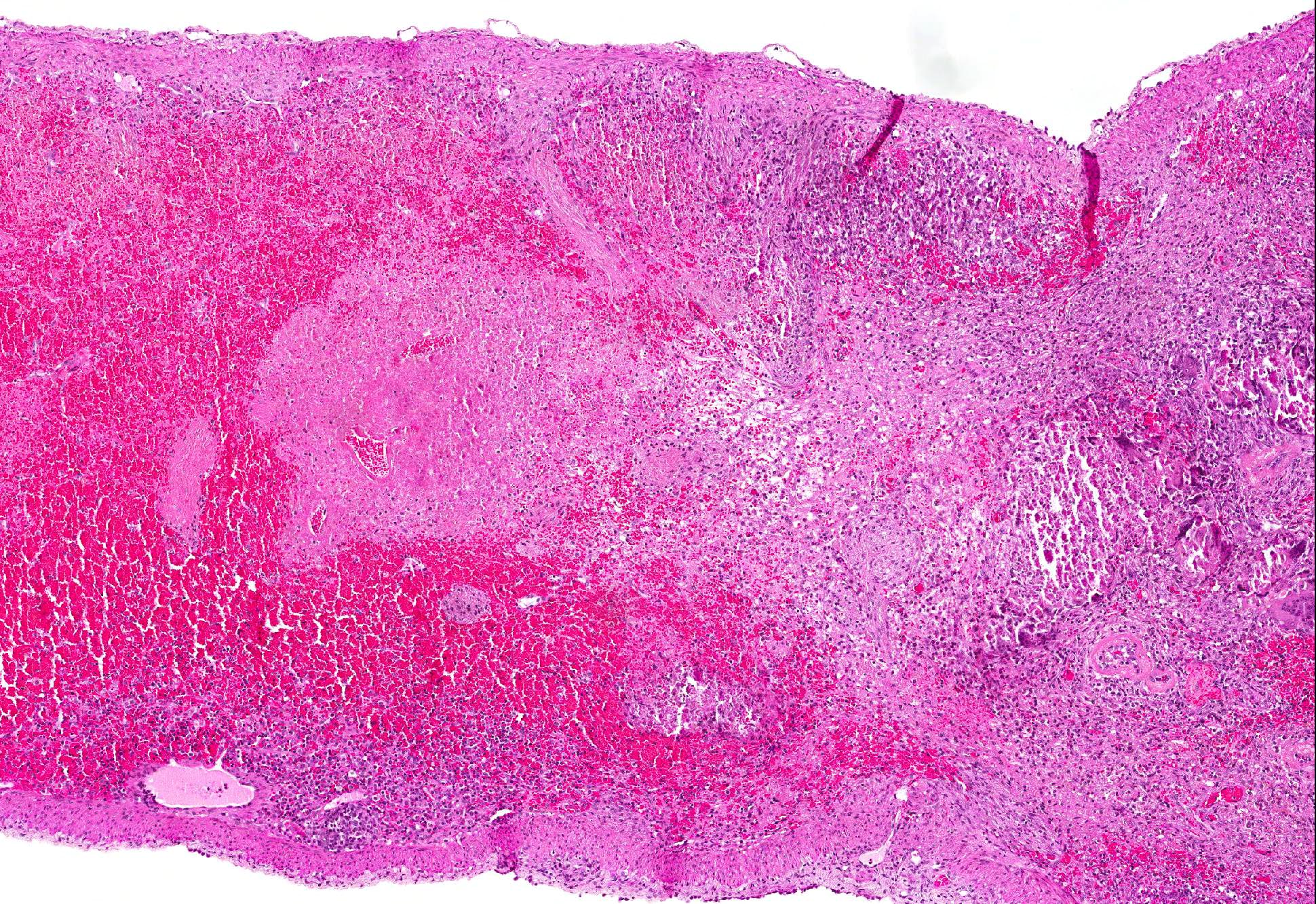
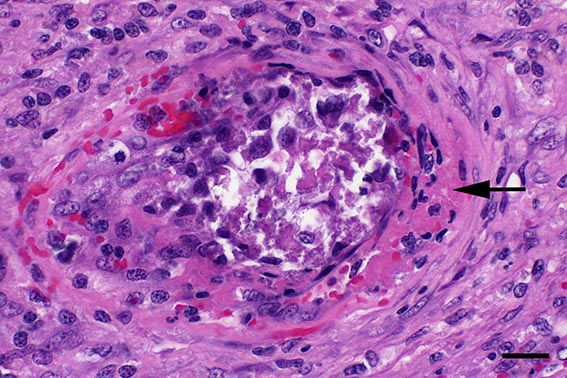
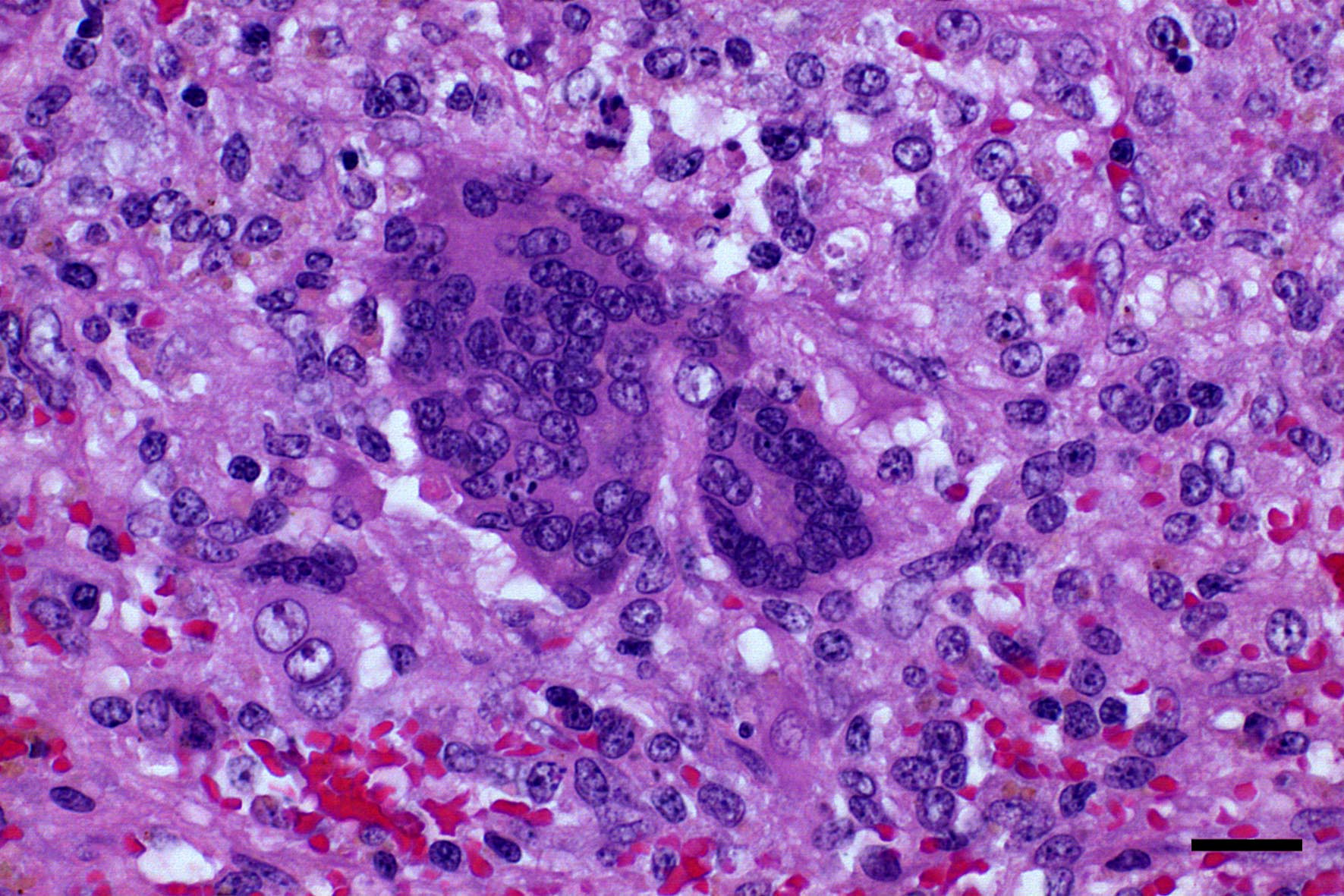
Microscopic Description: Spleen: Located at the acute angle and affecting 10-20% of the splenic parenchyma is a wedge-shaped area displaying diffuse, pale, eosinophilic, shadow-like histoarchitecture and a peripheral zone of moderate extravascular erythrocyte accumulation (acute hemorrhage) which extends into the adjacent normal tissue. The stromal and parenchymal cells display ill-defined cellular borders, cytoplasmic hypereosinophilic condensation, nuclear loss, pyknosis and karyorrhexis (coagulation necrosis). There is minor multifocal basophilic granular change of the necrotic debris (dystrophic mineralization). At the border to the normal appearing tissue are oligofocal medium sized arteries with pyknotic, karyorrhectic and lost endothelia and smooth muscular cells, intramural deposition of an hypereosinophilic amorphous mass (fibrin) and minor and variable infiltration by degenerating neutrophils, macrophages and foreign body-type and Langhans-type multinucleated giant cells (fibrinonecrotizing vasculitis). The lumen of multiple affected arteries is occluded by an eosinophilic fibrillary to amorphous mass with scant encased degenerating neutrophils and macrophages (fibrin-rich thrombus). The white pulp in the adjacent parenchyma is of mildly reduced cellularity and displays a mild loss of small differentiated lymphocytes with heterochromatic nuclei (lymphoid depletion) combined with a relative increase in moderately sized lymphoblastic cells with round, euchromatic nucleus, multiple mitotic figures and a mildly increased number of tingible body macrophages. The serosal mesothelial cells are multifocally enlarged and display round euchromatic nuclei (reactive change).
Contributors Morphologic Diagnosis:
Spleen: Vasculitis, fibrinonecrotizing, oligofocal, acute, moderate, with multinucleated giant cells, arterial thrombosis, necrosis and hemorrhage (infarct).
Contributors Comment: This case of moderate, acute, oligofocal, fibrinonecrotizing vasculitis with multinucleated giant cells, arterial thrombosis, necrosis and hemorrhage is the histopathological correlate of the grossly detected anemic infarcts at the acute angle of the spleen. These infarcts at the acute angle of the spleen (there is a special German term for that: Milzrandinfarkte) are considered as one of the most characteristic if not pathognomonic lesions present in 1 87% of cases of classical swine fever (CSF; synonym: hog cholera).13
This case from an experimental study was selected for the Wednesday Slide Conference because in contrast to most classic textbooks of veterinary pathology including Jubb, Kennedy and Palmers Pathology of Domestic Animals and Pathologic Basis of Veterinary Disease which describe hemorrhagic infarcts in CSF,13,14 the current case demonstrates ischemic core infarct areas, suggestive of a very early stage of an infarct pathogenesis. Furthermore, the multinucleated giant cells observed in this case are a rare feature, possibly due to a marked activation of monocytes / macrophages, which is a key element of CSF pathogenesis.4,8
Etiology:
The etiologic agent of CSF is the classical swine fever virus (CSFV), a pestivirus of the flaviviridae family. It is related to the other well-known species of the genus pestivirus bovine virus diarrhea virus-1 and -2, border disease virus as well as novel pestiviruses including HoBi-like viruses (atypical pestivirus) and Bungowannah virus.12 Notably, some of the related ruminant pestiviruses including bovine virus diarrhea virus can occasionally infect pigs,11 leading to cross-reactive antibodies and false positive serological results.5 In contrast, evidence of naturally occurring CSFV-infection in ruminants or other species is lacking. The CSFV is a spherical, enveloped, single-stranded, positive-sense RNA virus with a diameter of ~ 50 nm. The ~12.3 kb RNA consists of a single open reading frame encoding the four structural proteins C, Erns, E1, and E2, as well as the non-structural proteins Npro, p7, (NS2-3), NS2, NS3, NS4A, NS4B, NS5A, and NS5B.12
Epidemiology:
CSF is one of the most important diseases of domestic pigs and notifiable to the World Organization for Animal Health. The main hosts for CSFV are domestic pigs (Sus scrofa domesticus) and European wild boars (Sus scrofa scrofa). Furthermore, it has been proven experimentally that warthogs (Phacochoerus africanus) and bushpigs (Potamochoerus larvatus) are also susceptible to CSFV.3 Currently CSF is endemic in South and Central America, parts of Eastern Europe, and Asia and represents a constant threat for all other pig producing countries.1
CSFV can be transmitted horizontally, mainly by the oronasal route. Furthermore, vertical in utero transmission is possible. The normal incubation time is four to seven days. Diseased pigs are highly viremic and shed virus at least from the beginning of clinical disease until death or the occurrence of neutralizing antibodies. Virus is shed in saliva, lacrimal secretions, urine, feces and semen.1
Clinical Course:
Depending on the virulence of the CSFV strain and the age and constitution of the host the clinical course of CSF can be peracute, acute or chronic. In the classical acute form, initial atypical clinical signs such as high fever, anorexia, gastrointestinal symptoms, weakness and conjunctivitis progress to the characteristic hemorrhagic fever and neurological symptoms with a mortality of up to 100% after 2 to 4 weeks. However, a variable proportion of pigs may not progress and recover from the disease. In the chronic form clinical symptoms such as remitting fever, depression, and wasting are usually non-specific and inevitably lead to death after a disease duration of 1 - 3 months. Furthermore, in utero infection is a special situation which leads to fetal death, resorption, abortion, mummification, stillbirth, malformations or birth of persistently infected piglets which show runting, late onset disease and inevitably death after 2 11 months.1
Macroscopic Changes:
Pigs may succumb to peracute CSF without any characteristic gross lesions. Characteristic macroscopic findings in acute CSF are dermal erythema and cyanosis at the margins of the ears, limbs and ventral abdomen. Petechial and ecchymotic hemorrhages can be observed in skin, larynx, epiglottis, urinary bladder, gastric mucosa, and the serosal surfaces of kidneys, lungs, and heart. The lymph nodes can present a typical marbled appearance due to hemorrhages with blood in the peripheral sinuses. Furthermore, there is necrotizing to suppurative tonsillitis, infarcts at the margin of the spleen, as well as hydropericardium, hydrothorax, and hydroperitoneum. In chronic CSF lesions include atrophic lymphatic organs, splenic infarcts, necrosis of gut associated lymphoid tissues including colonic button ulcers. Furthermore, due to the immunosuppression a magnitude of secondary and non-specific lesions can occur. Gross lesions in late onset disease in persistently infected pigs resemble those of the chronic form. Typical malformations in piglets include cerebellar hypoplasia, thymic atrophy, and deformities of the head and limbs.1,13,14
Pathohistologic Changes:
The most important pathohistological findings in acute CSF are multifocal necrotizing vasculitis and lymphoid necrosis and depletion in the lymphatic organs. Vasculitis is often followed by thrombosis, infarction and hemorrhages. In addition to the characteristic gross splenic infarcts, infarcts can also develop in lymph nodes, skin, tonsil, gallbladder, and large intestine. No matter if neurological clinical symptoms have been reported or not, the brain is among the best tissues to check for histological changes. They present as lymphocytic panencephalitis with marked perivascular cuffing and extravasation of blood plasma protein. Chronic cases may also present with mesangioproliferative glomerulonephritis. In utero infected piglets can be affected by hypomyelination inducing congenital tremors. Furthermore, the physeal growth plates may exhibit zones of persistent primary spongiosa (growth arrest lines) presumably reflecting viral destruction of osteoclasts.13
Pathogenesis:
Infection usually occurs via the oronasal route and the CSFV enters the body mainly via the epithelial cells or M-cells of the tonsillar crypts. Primary replication takes place within macrophages in the tonsils and local oropharyngeal lymph nodes, followed by mononuclear cell-associated viremia mainly to other lymphoid organs including spleen, thymus, lymph nodes and mucosa associated lymphoid tissues.14 Furthermore, CSFV can also be found in variable amounts in all other organs of the body including skeletal muscles, heart, lung, liver, pancreas, stomach, duodenum, jejunum, ileum, kidney, urinary bladder, brain, spinal cord.6 The CSFV envelope glycoproteins Erns and E2 are responsible for attachment to the putative virus receptor CD46 together with heparan sulfates.2 The most important host cells are macrophages and dendritic cells, but CSF can also be found in endothelial cells and various epithelial cells, lymphocytes and megakaryocytes. Although activation of macrophages is a well-known feature of CSF, multinucleated giant macrophages as observed in this case are not described.4,8 Especially the infection of plasmacytoid dendritic cells induces secretion of large quantities of interferon ?, which is a key element in the pathogenesis of CSF. The high serum level of interferon ? is suggested to be the central inducer of immune cell dysregulation, which manifests as lymphocyte apoptosis, depletion and immunosuppression as well as bone marrow suppression.10 Furthermore, a marked increase in macrophage / monocyte-derived pro-inflammatory cytokines such as TNF ?, IL-1, and IL-6 is suggested to be the main mediator of systemic endotheliotoxicity. Cytokine induced activation of endothelial cells represents the basis for light microscopically visible endothelial swelling. This is followed by degeneration and necrosis of endothelial cells inducing breakdown of the endothelial barrier, fibrinonecrotizing vasculitis, activation of the clotting system, edema and hemorrhages.14 The formation of arterial infarcts at the multifocal sites of vasculitis within the spleen leads to vascular occlusion followed by ischemic necrotic cell death of the dependent parenchyma. The anatomy of the spleen with single blood supply and minimal anastomoses is an important predisposing factor for infarction.7 Although it is also most possibly related to some microanatomical features, it is still enigmatic why the infarcts are aligned along the acute angle of the spleen in CSF but not in other diseases with splenic infarcts such as African swine fever, highly-pathogenic porcine reproductive and respiratory syndrome and chronic erysipelas.9 As can be seen in the current case, the infarcted areas initially can be pale due to the lack of blood inflow following complete arterial occlusion. This is rapidly followed by hemorrhage from damaged vessels and inflow of blood from the surrounding parenchyma with intact perfusion leading to the commonly illustrated hemorrhagic infarcts. Secondary pallor due to cell swelling, hemoglobin degradation and diffusion within the affected area usually does not take place in the spleen due to its spongy consistency.7
Differential Diagnosis:
A good overview concerning the differential diagnosis of porcine hemorrhagic fevers is presented in Sánchez-Vizcaíno et al. (2015).9 Concerning splenic infarction in pigs, the most important etiologic differential diagnoses are CSF, African swine fever, highly-pathogenic porcine reproductive and respiratory syndrome, and septic and embolic infarction from other primary lesion sites such as endocarditis valvularis thromboticans in chronic erysipelas.
JPC Diagnosis: 1. Spleen, red pulp: Vasculitis, necrotizing, multifocal to coalescing with multifocal infarcts, Sus scrofa domesticus, porcine.
2. Spleen, white pulp: Lymphoid depletion, diffuse, severe.
Conference Comment: The contributor provides a complete review of classical swine fever. The multinucleated giant cells noted by the contributor were seen by conference participants as well. It is possible, that as the moderator suggests, these cells form because monocytes and macrophages are markedly activated as part of the CSF pathogenesis. Attendees debated this concept and pointed out that pestiviruses do not have a fusion protein and should not inherently cause fusion of affected cells.13 Although the process of multinucleated giant cell formation in inflammation is not well understood it does require that macrophages be bathed in cytokines like IFN-?, IL-3, IL-4, IL-13 and GM-CSF. Subsequently, membranes of adjacent macrophages express fusogenic proteins such as: DC-STAMP, ?1 and ?2 integrins, CD44, CD47, macrophage fusion receptor, fusion regulator protein (FRP-1; CD98), and P2X7 (ligand gated ion channel activated by ATP that forms a pore).1
Friedrich-Loeffler-Institut
Federal Research Institute for Animal Health
Department of Experimental Animal Facilities and Biorisk Management
Südufer 10, 17493 Greifswald Insel Riems, Germany.
References:
1. Blome S, Staubach C, Henke J, Carlson J, Beer M. Classical swine fever-an updated review. Viruses 2017:9(4).
2. Drager C, Beer M, Blome S. Porcine complement regulatory protein CD46 and heparan sulfates are the major factors for classical swine fever virus attachment in vitro. Arch Virol 2015:160(3):739-746.
3. Everett H, Crooke H, Gurrala R, Dwarka R, Kim J, Botha B, et al. Experimental infection of common warthogs (Phacochoerus africanus) and bushpigs (Potamochoerus larvatus) with classical swine fever virus. I: Susceptibility and transmission. Transbound Emerg Dis 2011:58(2):128-134.
4. Gomez-Villamandos JC, Ruiz-Villamor E, Bautista MJ, Sanchez CP, Sanchez-Cordon PJ, Salguero FJ, et al. Morphological and immunohistochemical changes in splenic macrophages of pigs infected with classical swine fever. J Comp Pathol 2001:125(2-3):98-109.
5. Hoffmann B, Beer M, Schelp C, Schirrmeier H, Depner K. Validation of a real-time RT-PCR assay for sensitive and specific detection of classical swine fever. J Virol Methods 2005:130(1-2):36-44.
6. Liu J, Fan XZ, Wang Q, Xu L, Zhao QZ, Huang W, et al. Dynamic distribution and tissue tropism of classical swine fever virus in experimentally infected pigs. Virol J 2011:8:201.
7. Mosier DA: Vascular Disorders and Thrombosis. In: Zachary JF, ed. Pathologic Basis of Veterinary Disease. 6th ed. St. Louis, Missouri: Elsevier; 2017: 44-72.
8. Sanchez-Cordon PJ, Nunez A, Salguero FJ, Pedrera M, Fernandez de Marco M, Gomez-Villamandos JC. Lymphocyte apoptosis and thrombocytopenia in spleen during classical swine fever: role of macrophages and cytokines. Vet Pathol 2005:42(4):477-488.
9. Sanchez-Vizcaino JM, Mur L, Gomez-Villamandos JC, Carrasco L. An update on the epidemiology and pathology of African swine fever. J Comp Pathol 2015:152(1):9-21.
10. Summerfield A, Ruggli N. Immune responses against classical swine fever virus: between ignorance and lunacy. Front Vet Sci 2015:2-10.
11. Tao J, Liao J, Wang Y, Zhang X, Wang J, Zhu G. Bovine viral diarrhea virus (BVDV) infections in pigs. Vet Microbiol 2013:165(3-4):185-189.
12. Tautz N, Tews BA, Meyers G. The molecular biology of pestiviruses. Adv Virus Res 2015:93:47-160.
13. Valli VEO, Kiupel M, Bienzle D. Hematopoietic system. In: Maxie MG, ed. Jubb, Kennedy and Palmer"s Pathology of Domestic Animals. Vol 3. 6th ed. St. Louis, Missouri: Elsevier; 2016: 102-267.
14. Zachary JF. Mechanisms of microbial infections. In: Zachary JF, ed. Pathologic Basis of Veterinary Disease. 6th ed. St. Louis, Missouri: Elsevier; 2017: 132-241.