Joint Pathology Center
Veterinary Pathology Services
Wednesday Slide Conference
2018-2019
Conference 12
12 December, 2018
CASE III 111474 (JPC 4084217)
Signalment: 7-year-old, male castrated, Shih Tzu dog (Canis familiaris)
History: A 7-year-old, male castrated, Shih Tzu dog was referred to the Cornell University Hospital for Animals for acute neurologic signs after 4 episodes of vomiting. The dog had a 4-5 year history of polyuria and polydipsia (PU/PD).
After vomiting overnight, the following day the dog showed signs of lethargy and ataxia which progressed to disorientation and lateral recumbency. The dog was brought to the referring veterinarian where blood work showed > 180 mmol/μl of sodium [144-160 mmol/μl]. Initial fluid therapy consisting of lactated Ringer's solution (LRS) was administered (unknown dose); however, the dog began head pressing. Furosemide (5 mg/kg) was administered. Soon after the patient had a focal seizure and received an additional 4 mg/kg of furosemide. The dog was referred to Cornell University Hospital for Animals’ Emergency Service. He had an additional seizure during transit.
On examination, the patient was stuporous, hyperthermic (104.6 F), and tachycardic (200 bpm) with a weak femoral pulse. The patient was approximately 9% dehydrated and had tacky, injected mucous membranes with a capillary refill time of 1 second. The initial blood pressure was 97/24 with a mean of 43. Anisocoria was present (meiosis OD). A neurologic consultation revealed a forebrain neurolocalization. The patient was recumbent with an absent menace response and nasal sensation, bilaterally. Proprioception was absent in all four limbs with intact spinal reflexes. No head or neck pain was noted.
Point of care blood work (PCV, total solids and venous blood gas) revealed severe hypernatremia (182 mEq/L; 140–154 mEq/L) and hyperchloridemia (134 mEq/L; 104–119 mEq/L) with normokalemia (3.04 mEq/L; 3.8–5.4 mEq/L).
The patient was started on a custom fluid containing 175 mEq/L of sodium. A recheck of venous blood gas analysis revealed sodium of 191mEq/L. On the bases of the history, physical examination findings, and biochemical abnormalities, central diabetes insipidus was suspected. Fluid therapy was changed to a hyponatremic solution and with continued fluid adjustments and subcutaneous administration of 1mcg/kg of desmopressin, the serum sodium decreased to 177.7 mEq/L over the course of 12 hours. The patient’s mental status remained unchanged with an additional seizure. MRI revealed bilateral grey matter swelling characterized by a reduced size of the subarachnoid space, effacement of the sulci and compression of the adjacent white matter tracts consistent with moderate, diffuse, cerebral edema, which was primarily cytotoxic.
The sodium level improved steadily over the 48 hours of hospitalization and fluid therapy and there was a transient improvement of mentation, but the patient became bradycardic and nonresponsive to atropine and experienced cardiac arrest. No cardiopulmonary resuscitation efforts were made at the request of the owner.
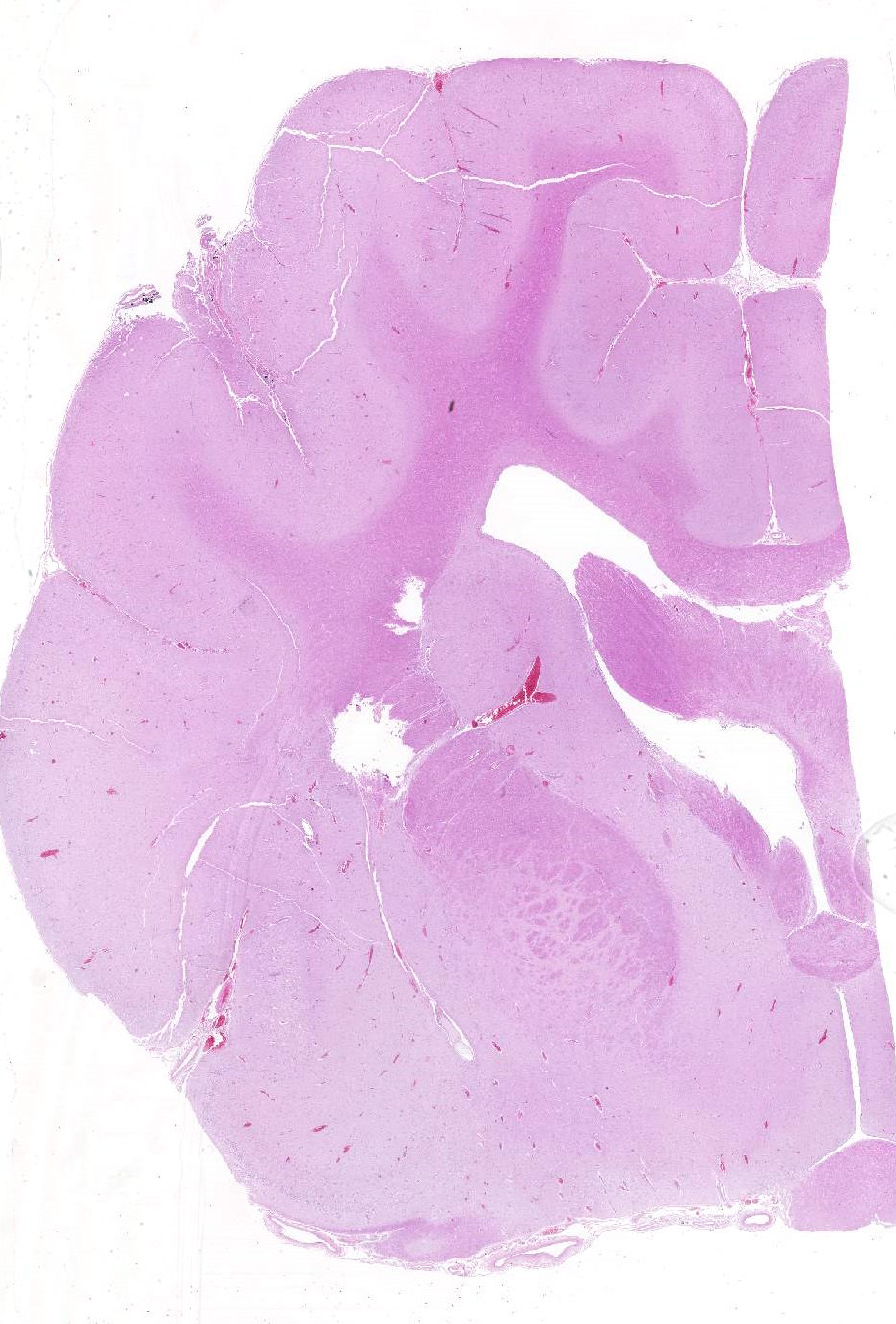
Gross Pathology: The submitting veterinarian performed the postmortem examination with no significant gross findings. He submitted fresh and fixed tissues for examination. Upon arrival at the diagnostic laboratory, the fresh tissues were severely autolyzed. In the fixed brain, there were light yellow areas of malacia in the cerebral gray matter that fluoresced when placed under an ultraviolet light with a wavelength of 365 nm.
Laboratory results: The sodium level of the brain tissue was 13,850 ppm (reference interval, calculated on a dry matter basis, 6000-8000 ppm).
Microscopic Description:
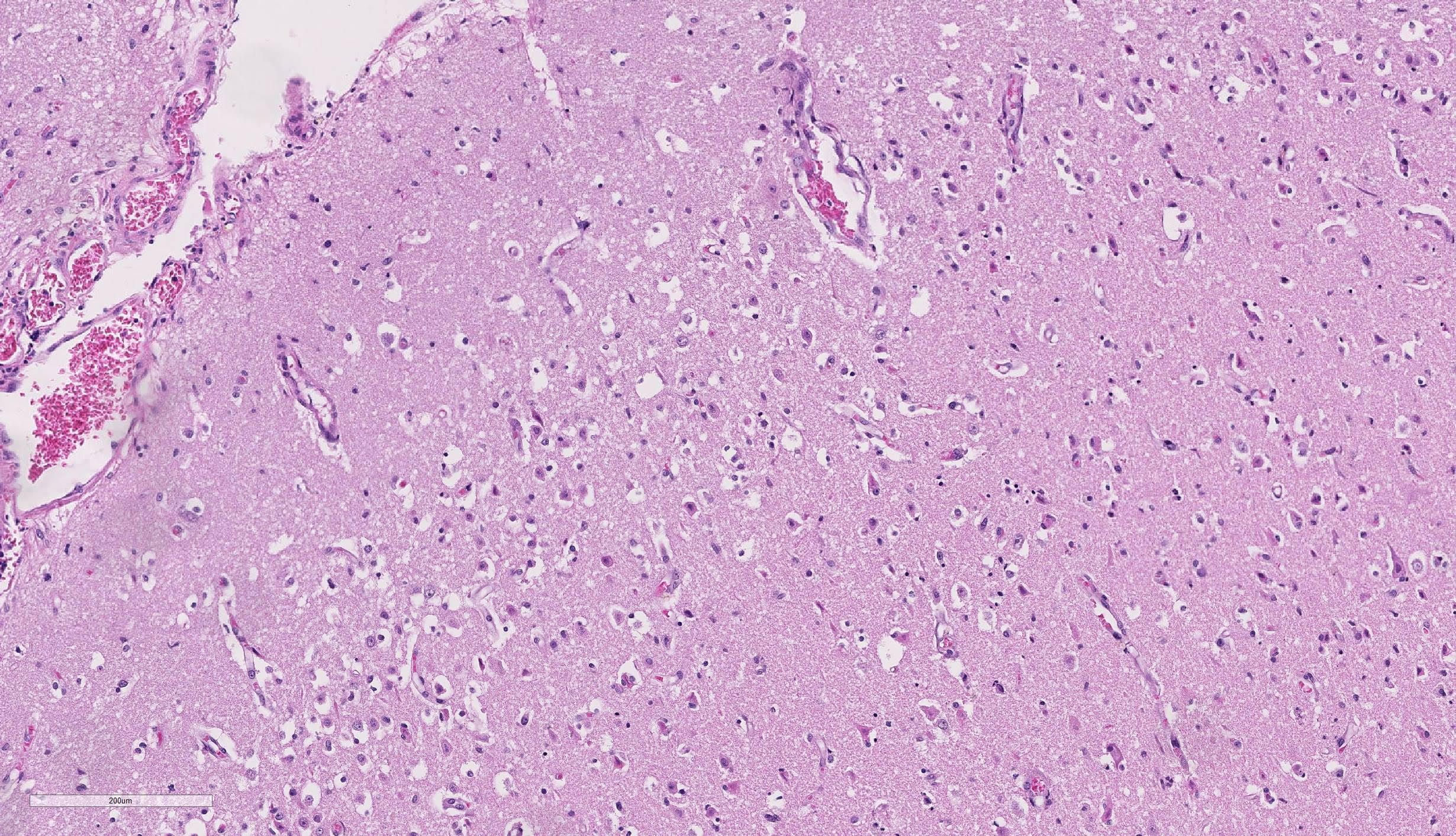
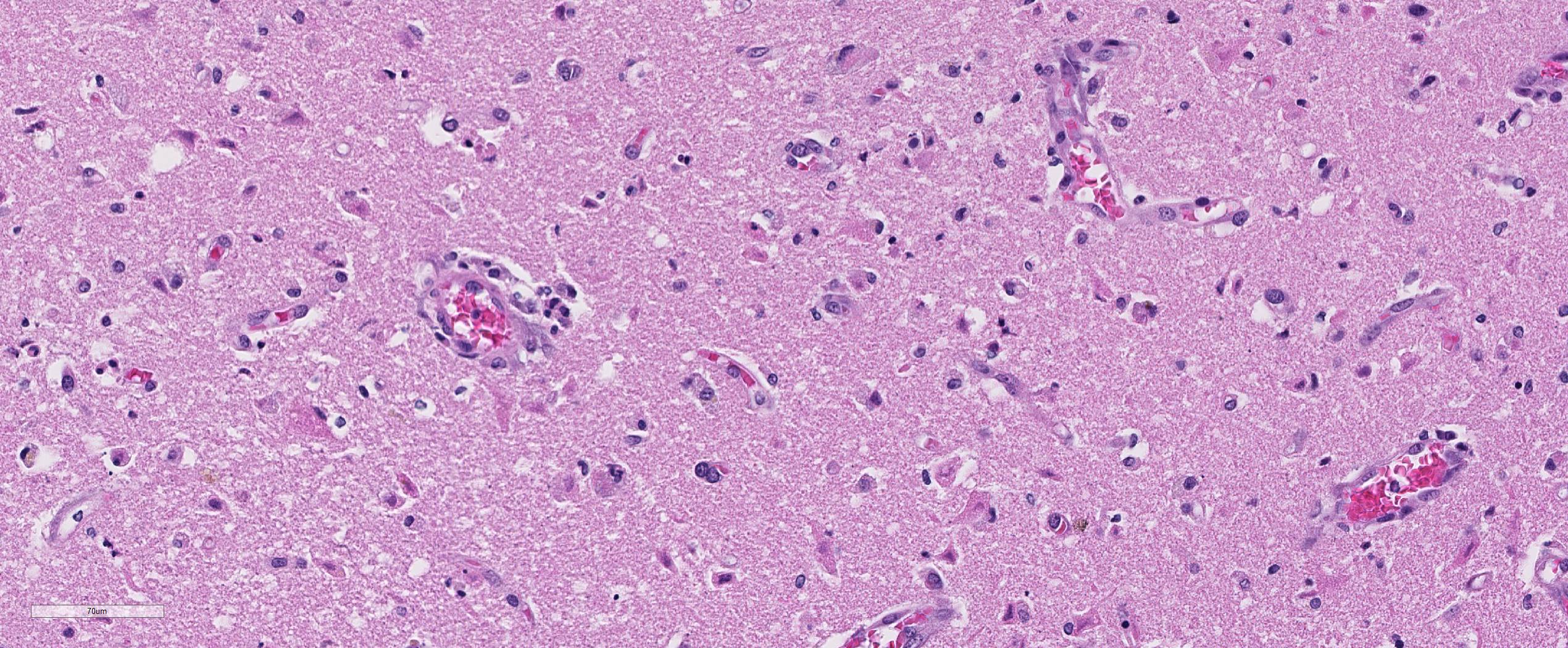
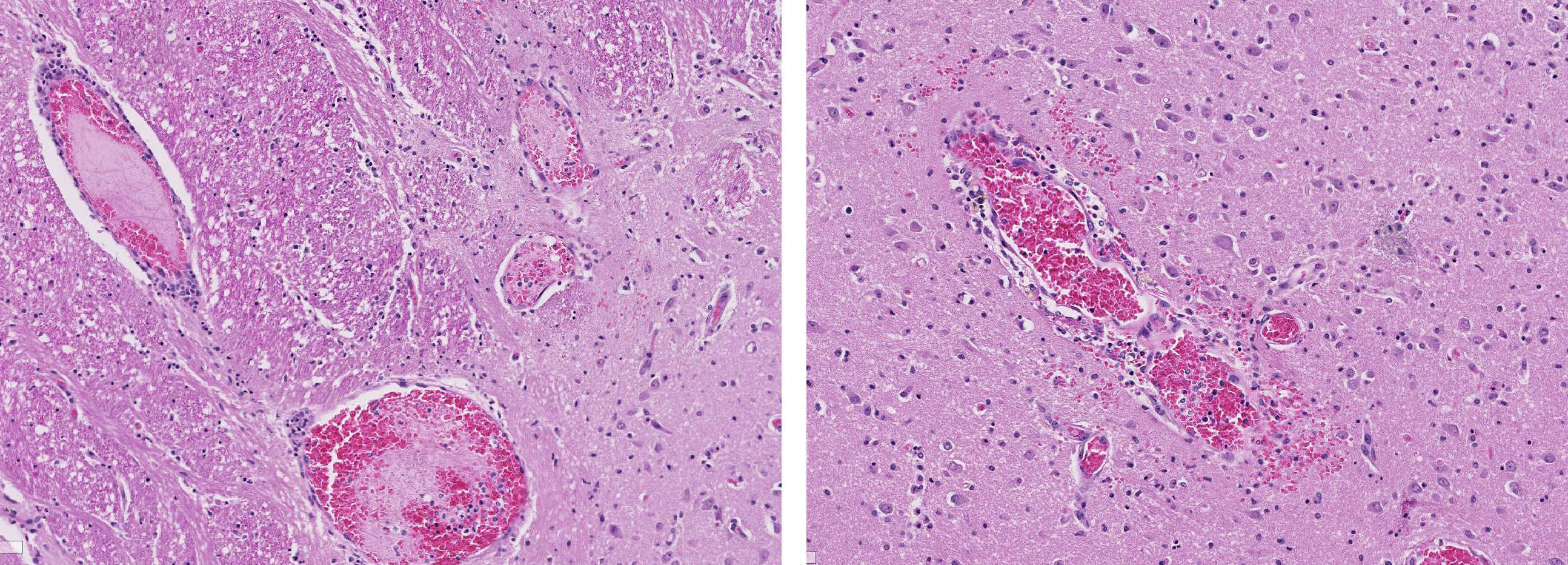
Brain, cerebral cortex: Diffusely, the blood vessels throughout all the sections are prominent, severely congested, lined by plump, reactive endothelial cells, and occasionally surrounded by small numbers of macrophages and fewer lymphocytes, hemorrhage, and fibrin within the Virchow-Robbin space. In the neuropil are scattered pyknotic and karyorrhectic cellular debris, most frequent in the mid to deep cerebral gray matter. In this area, there are frequent necrotic neurons: 1) angular with hypereosinophilic, variably microvacuolated cytoplasm that lack Nissl substance, with a smudgy nucleus without a nucleolus, or 2) appear as faint angular eosinophilic shadows of ghost neurons that lack a nucleus. The necrosis is most prominent in the mid to deep cerebral gray matter, while the perivascular cuffing, vascular branching, and perivascular fibrin and hemorrhage are most prominent in the caudate nucleus.
Pituitary gland (image not included): The pituitary gland is severely distorted by multifocal to coalescing cysts replacing and expanding 60% of the pars distalis, most of the pars intermedia, and 10% of the pars nervosa. The cysts are filled with variably eosinophilic, fibrillar to homogenous material, and lined by attenuated to low columnar, pseudostratified ciliated epithelium.
A variety of immunohistochemical and histochemical stains were applied to the brain sections. All positive and negative controls were adequate.
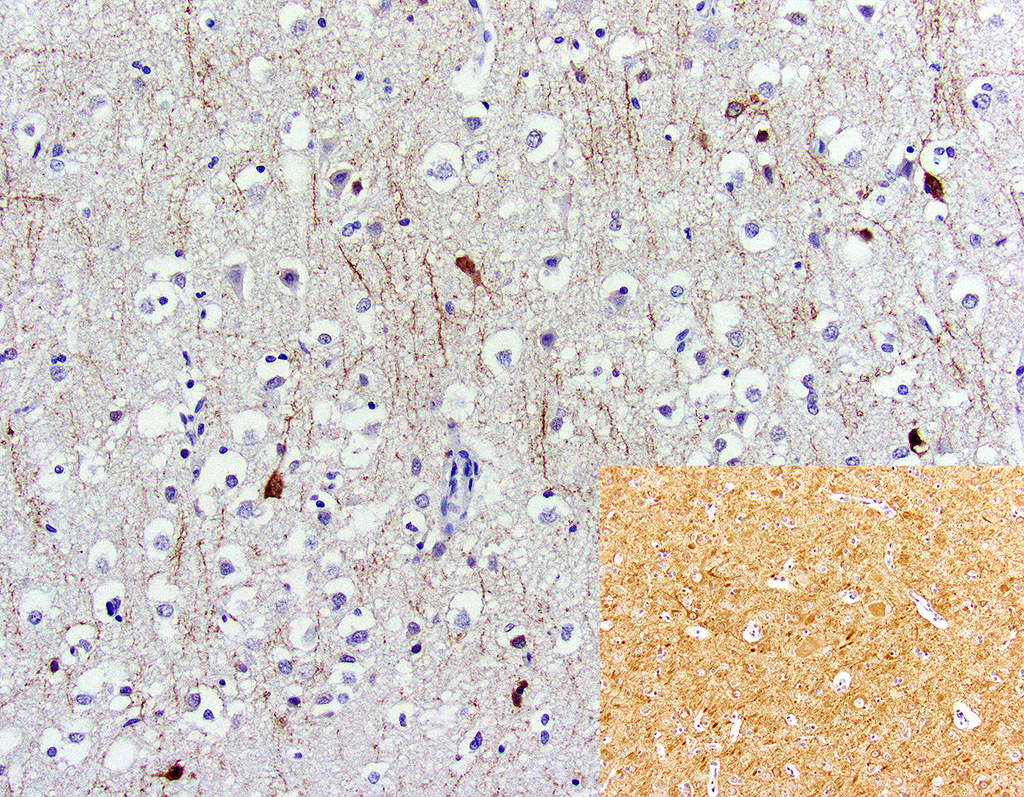
Microtubule associated protein 2 for neurons (MAP2) demonstrated strong but infrequent cytoplasmic immunoreactivity of the soma and moderate immunoreactivity of cytoplasmic processes in neurons of the mid to deep cerebral gray matter indicative of neuronal necrosis. An image of a positive control from a normal dog is included as an inset. In this image, the neuropil exhibits widespread moderate immunoreactivity with moderate to strong immunoreactivity of the neuronal soma and processes. The internal negative control is adequate highlighted by the negative glial cells and blood vessels.
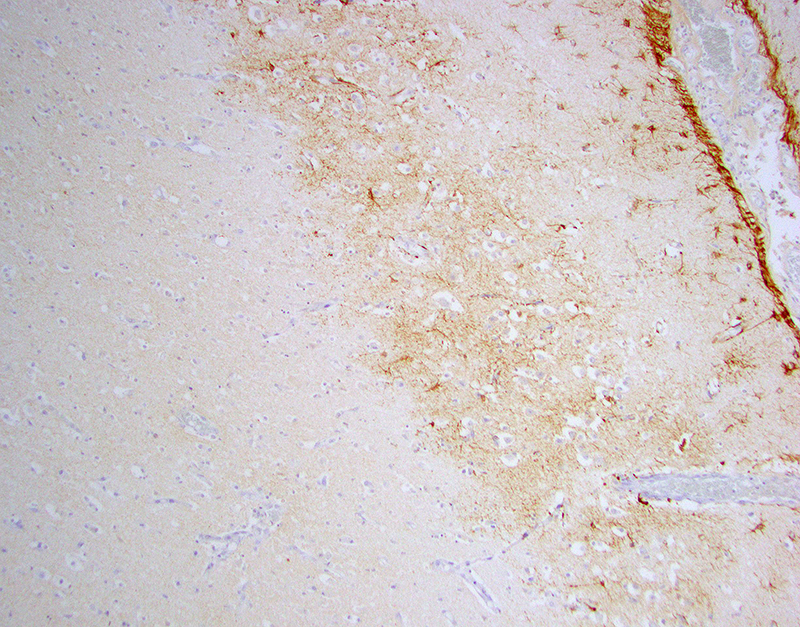
Glial fibrillary acidic protein (GFAP) for astrocytes highlights the well-demarcated cortical laminar necrosis affecting the mid to deep gray matter with very few remaining astrocytes in this area.
Olig2 for oligodendrocytes maps the GFAP in the mid to deep gray matter.
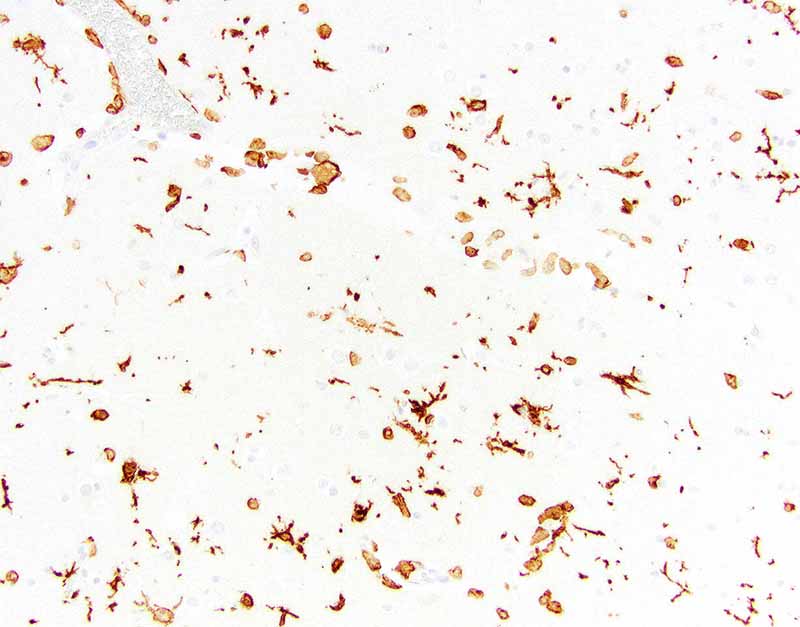
Ionized calcium binding adaptor molecule 1 (Iba1) for microglia and histiocytes in the affected mid to deep cerebral gray matter revealed three different populations of microglia . The first population is round, ameboid, or nonramified with strong cytoplasmic immunoreactivity interpreted as reactive microglia. The second population is stellate or ramified with strong cytoplasmic immunoreactivity interpreted as quiescent microglia. The third is a background population of small and round microglia and exhibits weak cytoplasmic immunoreactivity and were considered degenerate. Note perivascular mononuclear cells are predominantly macrophages.
Contributor’s Morphologic Diagnoses:
Brain: Severe, acute, cortical laminar necrosis with reactive microgliosis, reactive endothelium, histiocytic perivascular cuffing, perivascular hemorrhage and fibrin exudation
Contributor’s Comment: Hypernatremia is an infrequently encountered condition in dogs caused by decreased body fluid or sodium gain.14 Causes of hypernatremia include diabetes insipidus, insensible loss such as hyperventilation or fever14, lack of water intake due to hypothalamic defect resulting in a defective thirst response,13 iatrogenic administration of hypertonic saline or sodium bicarbonate or consumption of high salt content material,9 and decreased renal excretion of sodium due to hyperaldosteronism.3
In a recent retrospective study, the three most common causes of severe hypernatremia in dogs were central diabetes insipidus, alimentary loss, and fever/hyperthermia.14 Central diabetes insipidus (CDI) is characterized by diminished activity or lack of the antidiuretic hormone (ADH) due to damage of neuroanatomic sites involved in ADH secretion including the neurohypophysis, hypothalamic osmoreceptors, the supraoptic or paraventricular nuclei, or the superior portion of the supraopticohypophyseal tract.12 Underlying causes include vascular, autoimmune, infection, drugs, surgery, trauma, benign or metastatic pituitary-hypothalamic tumors. Patients with a normal thirst mechanism and free access to water generally do not present clinically; however, when water intake is restricted, hypernatremia occurs.1 In this case, the central diabetes insipidus was attributed to craniopharyngeal duct cysts and predisposed this dog to severe hypernatremia. However, given the widespread, severe lesions of the brain, the aggressive fluid therapy likely contributed to the severe histologic lesions.
Clinically, hypernatremia primarily affects the central nervous system and rapid changes in plasma sodium concentrations can cause severe, permanent, and lethal brain injury.2 The pathogenesis of these lesions is due to the failure to retain brain osmolarity. As the plasma osmolarity decreases with fluid therapy when these cases are recognized, the brain osmolarity fails to decrease at a similar rate due to failure of idiogenic osmoles to dissipate leading to cerebral edema.
Salt toxicity and water deprivation and its histologic features have been widely documented in other domestic animals including pigs, poultry, and ruminants.9 Most cases are caused by indirect salt poisoning which occurs when consumption of high levels of salt, followed by water deprivation, and then excessive water consumption occurs.9 Direct salt poisoning is in general rare, but primarily has been reported in cattle.15 The histopathologic lesions shared across these species include cerebral cortical laminar necrosis, perivascular mononuclear cuffing, and cerebral edema.8 Indirect salt poisoning in pigs is a well-known pathologic characterized by eosinophilic meningitis and perivascular eosinophilic cuffing.13
Neuropathologic alterations of hypernatremia in the dog have mostly been extrapolated from human and rodent studies with only brief descriptions in the literature. Most of the histologic description in canine cases has been quite variable. Cerebrocortical neuronal necrosis with symmetrical encephalomalacia of the brain stem has been reported in a dog diagnosed with pituitary-dependent hyperadrenocorticism that was euthanized due to hypernatremia 4 weeks post hypophysectomy.9 Neuronal degeneration and astrocytosis of the thalamus and hypothalamus has been reported in a young dog with hypodipsic hypernatremia.4 In a case of acute salt toxicity by ingestion of salt-flour mixture used as clay, diffuse white matter vacuolation with occasional perivascular cuffing by necrotic hyperchromatic mononuclear leukocytes with a thin ring of hemorrhage was observed with no evidence of axonal or neuronal necrosis.6
This case described the range of histologic changes that can be seen in dogs with hypernatremic encephalopathy which includes 1) cortical laminar necrosis, 2) reactive and necrotic endothelium with perivascular histiocytic cuffing, edema, fibrin, and hemorrhage, 3) reactive microgliosis, 4) white matter vacuolization, and 5) Purkinje cell necrosis. In the absence of relevant history, toxic-metabolic encephalopathies or global ischemic encephalopathy where widespread bilateral ischemic damage can occur in anesthetic deaths, cardiac arrest, severe anemia, hypotension, hypovolemic shock, and neonatal maladjustment syndrome may be considered. In this case, the sudden and aggressive ion alterations likely account for the brain lesions and the neurologic signs. In conclusion, severe hypernatremia should be considered as a differential in dogs with cortical laminar necrosis.
Contributing Institution:
Cornell University, College of Veterinary Medicine, Department of Biomedical Sciences, Section of Anatomic Pathology, Ithaca, New York, USA
http://www.vet.cornell.edu/biosci/pathology/
JPC Diagnosis: Cerebrum: Necrosis, cortical, laminar and segmental, multifocal to coalescing, with gliosis.
JPC Comment: The contributor provides an excellent review of hypernatremia in the dog. To add to the extensive list of differentials for polioencephalomalacia in the dog, other potential causes to be considered in the dog include lead, cyanide,thiamine deficiency, hypoglycemia, canine distemper infection, head trauma, cardiac arrest, and following portosystemic shunt ligation.8 Seizure activity has been documented to result in cortical damage in man and domestic and experimental animals, and the moderator discussed the possibility of this being the root cause of the laminar cortical necrosis in this animal.8
One of the classic symptoms of salt toxicity in affected animal species is blindness. In a previous WSC case of salt toxicity in a Vietnamese potbellied pig (WSC 2012-2013, Conference 21, Case 1), resulting from eating potato chips (this has been reported several times in the literature!), blindness was a long-term sequela following hospitalization and treatment and ultimately resulted in his euthanasia. While a number of changes occur in the brain following the development of hypernatremia and hyperosmolarity and may result in dysfunction of the optic pathway, equally severe and irreparable changes also result in the globe. Following ingestion of diet with 3% salt concentration (the minimum concentration associated with changes in the globe) for 3-4 days, the following ocular changes were noted in study pigs, rats, and rabbits – conjunctival injection, corneal and lens opacities, swelling of the optic disc, blindness, vitreous collapse, and ultimately progress to phthisis bulbi. Due to hyperosmolality, fluid loss from the vitreous resulted in vitreous shrinkage resulting in vascular damage, retinal and vitreous hemorrhage, posterior vitreal detachment, and retinal detachment.6 This suggests that while many of the CNS signs in salt-intoxicated animals may resolve with successful therapy to combat hyperosmolality, the changes in the eye may be irreversible and blindness, if present, permanent.
In the hospitalized dog and cat, hypernatremia is associated with a significant decrease in prognosis.13 Case fatality rates of dogs and cats with hypernatremia was 20.6 and 28.1% respectively compared to 4.4 and 4.5% with a normal blood or serum sodium concentration. In a review case histories in this study, 50% of the dogs and 38.5% of cats presented with hypernatremia, and the remaining animals developed hospital acquired hypernatremia.
References:
- Adrogue, H.J., and N.E. Madias. 2000. Hypernatremia. N Engl J Med 342: 1493-1499.
- Ayus, J.C., D.L. Armstrong, and A.I. Arieff. 1996. Effects of hypernatraemia in the central nervous system and its therapy in rats and rabbits. J Physiol 492 ( Pt 1): 243-255.
- Breitschwerdt, E.B., D.J. Meuten, C.L. Greenfield, L.W. Anson, C.S. Cook, and R.E. Fulghum. 1985. Idiopathic hyperaldosteronism in a dog. J Am Vet Med Assoc 187: 841-845.
- Crawford, M.A., M.D. Kittleson, and G.D. Fink. 1984. Hypernatremia and adipsia in a dog. J Am Vet Med Assoc 184: 818-821.
- Greco, D.S. 2012. Pituitary deficiencies. Top Companion Anim Med 27: 2-7.
- Heydarpour F, Derakhshandeh J, Fekn A, Heydarpour P. Salt poisoning blindness in Wistar rat, rabbit, and pig. Toxicol Environ Chem 2008; 90(5):1035-1042.
- Khanna, C., H.J. Boermans, and B. Wilcock. 1997. Fatal hypernatremia in a dog from salt ingestion. J Am Anim Hosp Assoc 33: 113-117.
- Mariana, CL, Platt SR, Newell SM, Terrell SP, Chrisman CL, Clemmons RM. Magnetic resonance imaging of cerebral cortical encrosis (polioencedcephalomalacia) in a dog.
- Maxie, M.G., editor. 2015. Jubb, Kennedy, and Palmer's Pathology of Domestic Animals. Elsevier, St. Louis, MO. 314-315 pp.
- Meij, B.P., G. Voorhout, T.S. van den Ingh, H.A. Hazewinkel, E. Teske, and A. Rijnberk.. Results of transsphenoidal hypophysectomy in 52 dogs with pituitary-dependent hyperadrenocorticism. Vet Surg 1998; 27: 246-261.
- Qureshi, S., S. Galiveeti, D.G. Bichet, and J. Roth. 2014. Diabetes insipidus: celebrating a century of vasopressin therapy. Endocrinology 155: 4605-4621.
- Shimokawa Miyama, T., E. Iwamoto, S. Umeki, M. Nakaichi, M. Okuda, and T. Mizuno. 2009. Magnetic resonance imaging and clinical findings in a miniature Schnauzer with hypodipsic hypernatremia. J Vet Med Sci 71: 1387-1391.
- Smith, D.L. 1957. Poisoning by sodium salt; a cause of eosinophilic meningoencephalitis in swine. Am J Vet Res 18: 825-850.
- Ueda, Y., K. Hopper, and S.E. Epstein. 2015. Incidence, severity and prognosis associated with hypernatremia in dogs and cats. J Vet Intern Med 29: 794-800.
- Van Leeuwen, J.A. 1999. Salt poisoning in beef cattle on coastal pasture on Prince Edward Island. Can Vet J 40: 347-348.