Signalment:
Gross Description:
Histopathologic Description:
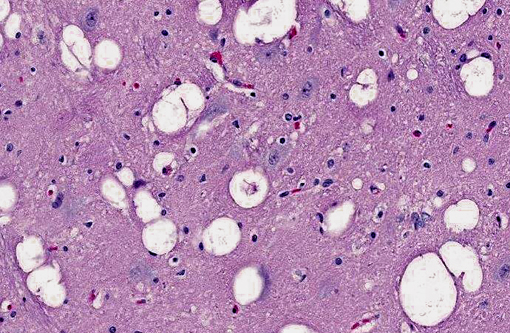
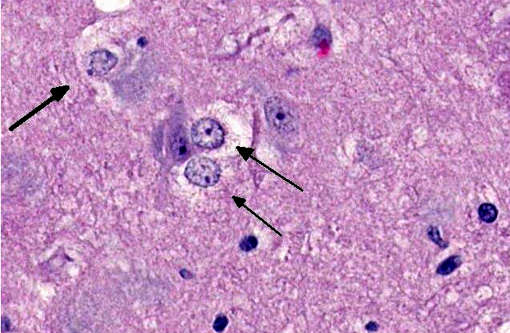
Brain: The gray matter and immediately adjacent white matter of the medulla, excluding the vagal and hypoglossal nuclei, contain numerous lacey, irregularly interconnected, unstained vacuoles that are well defined. At the edges of some affected areas astrocytes possess enlarged, pale homogeneous nuclei. The gray matter of the caudate nucleus, globus pallidus, peri-aqueductal gray matter, parts of the reticular formation, thalamus and hypothalamus are similarly affected, with much less change in the cerebrum and cerebellum.
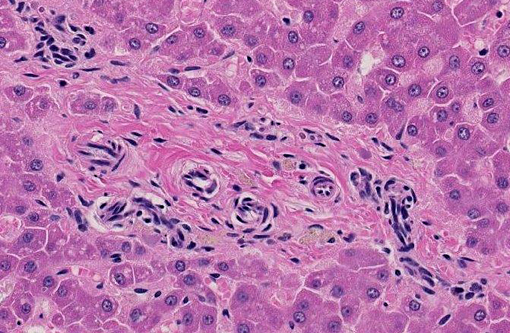
Liver: The hepatic lobules are mildly reduced in size. Although thin-walled veins are present in the largest portal areas near the capsule of the liver, none are apparent in the portal triads comprising the liver parenchyma.
Morphologic Diagnosis:
1. Brain: Spongiform encephalopathy, gray matter, with astrocytic nuclear hypertrophy (Alzheimer type II astrocytes).
2. Portal vein hypoplasia, liver.
Lab Results:
Condition:
Contributor Comment:
In acute liver failure, spongiform encephalopathy may lead to increased intracranial pressure. Studies suggest that intracranial hypertension complicates 25% of acute liver failure cases in humans but only 9% of those with subacute liver failure. In acute liver failure, an arterial ammonia concentration greater than 150 μmol/L predicts a greater likelihood of dying from brain herniation.(1) Spongiform change can be found in a variety of species, with the exception of the horse.Â
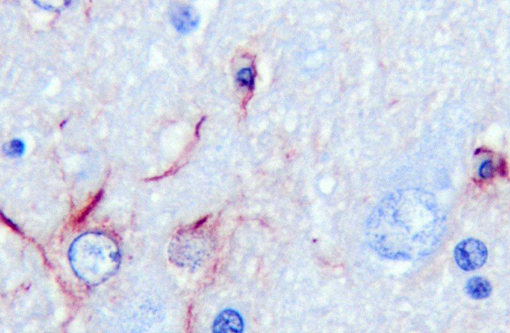
Of the compounds that escape detoxification by the failing liver, ammonia most easily crosses the blood-brain barrier. Although other compounds contribute, ammonia is accepted as the central cause of hepatoencephalopathy. It is considered that astrocyte dysfunction is central to ammonia toxicity in hepatoencephalopathy, and that functional neuronal changes are secondary. Astrocytic end processes surround capillaries in the CNS. Normally, this would ensure that any toxin entering the brain, such as ammonia, would be immediately metabolized, protecting other CNS cells from damage. In acute liver failure, increased brain ammonia concentrations affect transit of electrolytes and water across cell membranes, and cause astrocyte swelling, with cytotoxic brain edema. Primary alteration of the blood brain barrier results in vasogenic edema as well. Opening of the blood-brain barrier is thought to further exacerbate aberrant neuronal transmission and entry of toxins besides ammonia. Immunohistochemical staining with GFAP demonstrates some astrocytes with dense robust processes, while others near vacuoles stain poorly. CD18 reagent revealed multifocal clusters of large cells, with microglial morphology in affected tissue.Â
Astrocytes perform a number of other important metabolic supportive functions for neurons, including supplying them with GSH, the main cytosolic antioxidant.(2) Significant reduction in the activities of glutathione peroxidase and superoxide dismutase enzymes occur in the brain of rats exposed to ammonium acetate.(1)
Astrocytes also control GABAergic and glutamatergic activities at the synapse, especially terminating excitotoxic action in the synaptic cleft. Astrocytes take up glutamate, metabolizing it to glutamine that is transported back to excitatory neurons for neurotransmitter metabolism. In hepatoencephalopathy, there is correlation between astrocyte swelling and glutamine concentration. When the urea cycle is dysfunctional in the brain, ammonia is detoxified through its condensation with glutamate to form glutamine. Increased ammonia concentrations lead to mitochondrial permeability transition, and the loss of mitochondrial transmembrane potential.(1) Astrocytes also utilize glycolysis to produce lactate as a neuronal energy source.
There are additional roles for inflammation in hepatoencephalopathy. In advanced stages of acute liver failure, the brain produces a number of pro-inflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin (IL)-1β and IL-6.Â
JPC Diagnosis:
1. Liver: Venous hypoplasia, portal and central, diffuse, marked, with arteriolar hyperplasia, lymphangiectasia, and mild lobular and hepatocellular atrophy.Â
2. Cerebral cortex, gray matter: Spongiform change, multifocal to coalescing, marked, with Alzheimer type II astrocytosis and microgliosis.Â
Conference Comment:
The conference moderator led a review of the serum biochemistry abnormalities typical of PSS with hepatic atrophy. Fasting and postprandial serum bile acid concentrations are elevated for at least two reasons: 1) portal blood has a high concentration of bile acids, and bypasses the liver, which in health removes approximately 95% of bile acids from portal blood; and 2) hepatic atrophy that follows chronic shunting causes loss of hepatic functional mass, impairing the livers ability to clear bile acids from the blood. Hyperammonemia, as discussed by the contributor, is also usually present in PSS because ammonia bypasses the liver, where hepatic urea cycle enzymes catalyze its conversion to urea. Consequently, animals with PSS may develop ammonium biurate crystalluria, as ammonium biurate precipitates in alkaline urine. With chronicity and the development of hepatic atrophy, loss of hepatic functional mass may result in hypoalbuminemia, hypoglycemia, hypocholesterolemia and/or decreased blood urea nitrogen (BUN) concentrations. Levels of cholestatic markers (e.g. alkaline phosphatase, gamma glutamyltransferase) and hepatocellular leakage enzymes (e.g. alanine transaminase, aspartate transaminase, sorbitol dehydrogenase, glutamate dehydrogenase) are usually within reference intervals. Animals with PSS may have microcytic anemia of unknown cause.(4)
References:
2. Sidoryk-Wegrzynowicz M, Wegrzynowicz M, Lee E. Bowman AB, Aschner M. Role of astrocytes in brain function and disease. Toxicol Pathol. 2011;13:115-123.
3. Stalker MJ, Hayes MA. Liver and biliary system. In: Maxie MG, ed. Jubb, Kennedy and Palmers Pathology of Domestic Animals. 5th ed. Philadelphia, PA: Elsevier Saunders; 2007:2:302-304.
4. Bain PJ. Liver. In: Latimer KS, ed. Duncan & Prasses Veterinary Laboratory Medicine Clinical Pathology. 5th ed. Ames, Iowa: Wiley-Blackwell; 2011:227, 415-417.