WSC 23-24, Conference 9, Case I:
Signalment:
8-year-old, male neutered Pembroke Welsh corgi, canine (Canis lupus familiaris)
History:
This dog developed labored breathing. An echocardiogram showed pulmonary hypertension and degenerative valve disease of the mitral, tricuspid, and pulmonic valves. The dog was started on sildenafil and pimobendan therapy and improved; however, approximately 1.5 months after the initial diagnosis the dog went into decompensated congestive heart failure and was humanely euthanized.
Gross Pathology:
The lungs were soft, wet, and mottled pink and reddish brown. The right ventricle of the heart was moderately dilated with a thickened wall. Both atrioventricular valves had moderate endocardiosis. The liver had evidence of chronic passive congestion (moderate enlargement, rounded lobar margins, meaty texture, roughened surface, and small amounts of fibrin on the capsular surface). The dog had mild ascites.
Microscopic Description:
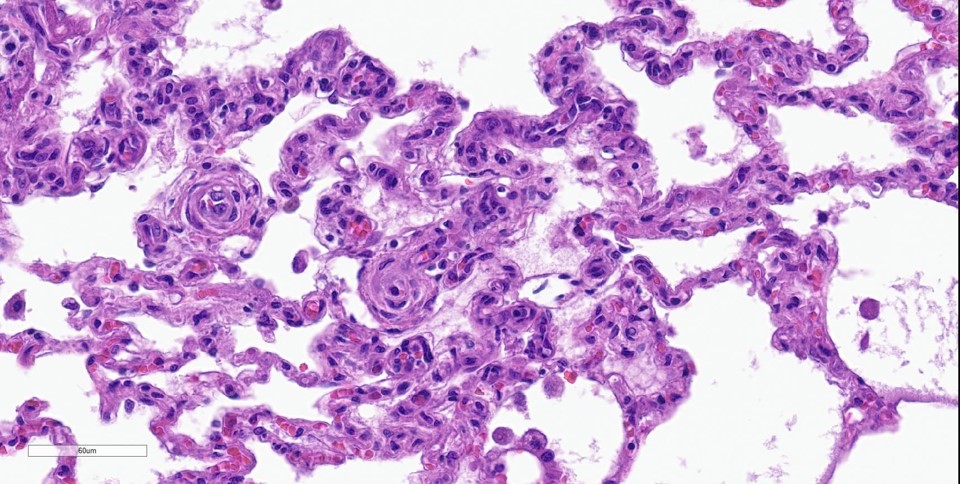
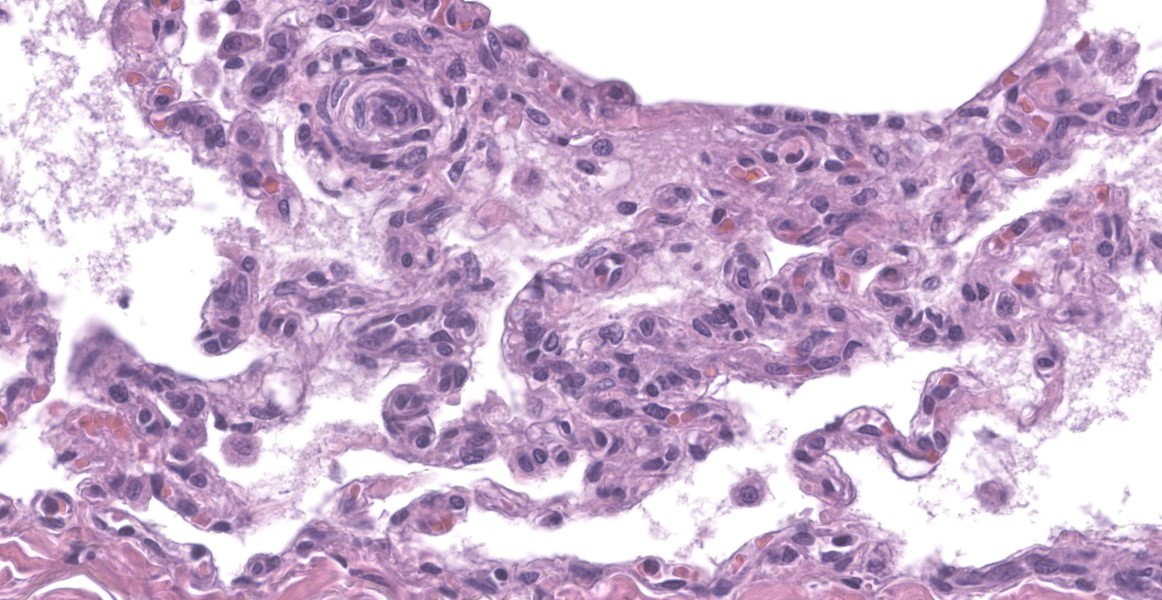
Lung: Throughout all sections there are multiple variably sized, poorly demarcated areas within which there is increased cellularity of alveolar septa and lumina. Within these areas, alveolar septa are mildly to moderately expanded by variable numbers of mesenchymal cells and rare small amounts of collagen. In a few small areas, the expansion of this mesenchymal population results in markedly thickened and tortuous alveolar septa which obscure the adjacent alveolar lumina. There is frequent, regionally variable, type II pneumocyte hyperplasia with a few small areas containing markedly hypertrophied, vacuolated, and/or misshapen type II pneumocytes with up to 3 nuclei. Alveolar spaces contain large numbers of macrophages which infrequently contain finely granular, golden-brown, intracytoplasmic pigment (hemosiderin) as well as small numbers of neutrophils and lymphocytes. Infrequently, alveolar
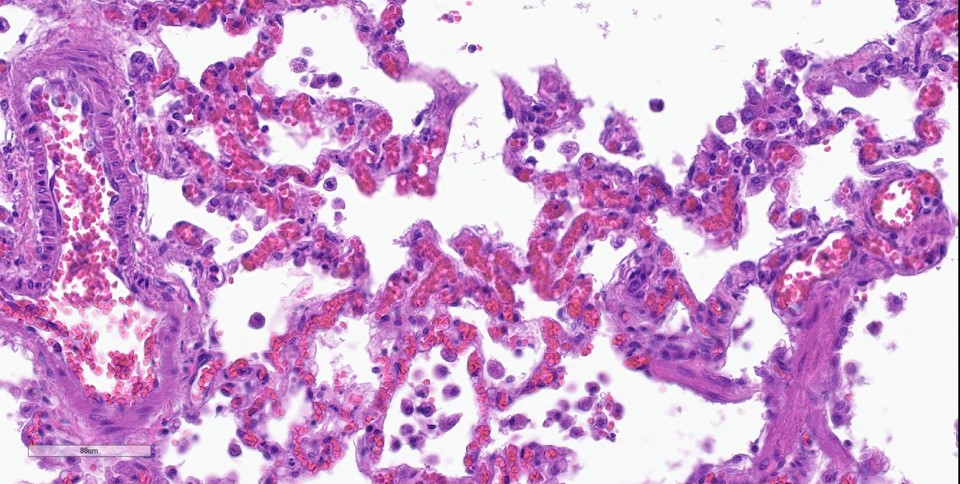
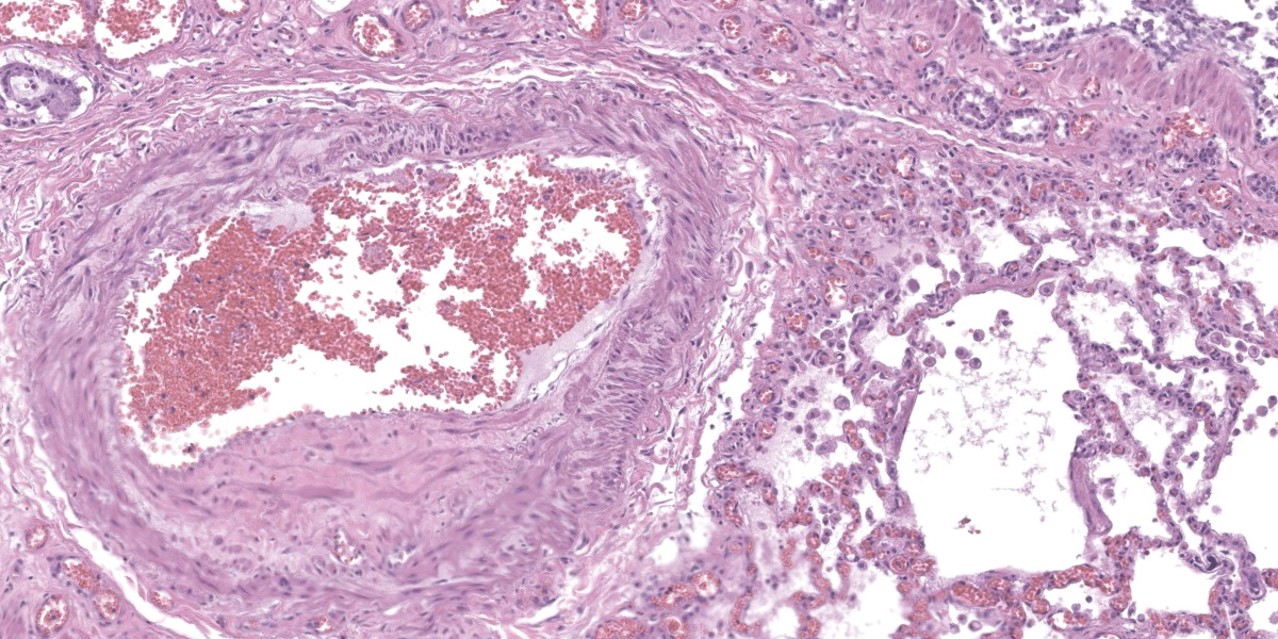
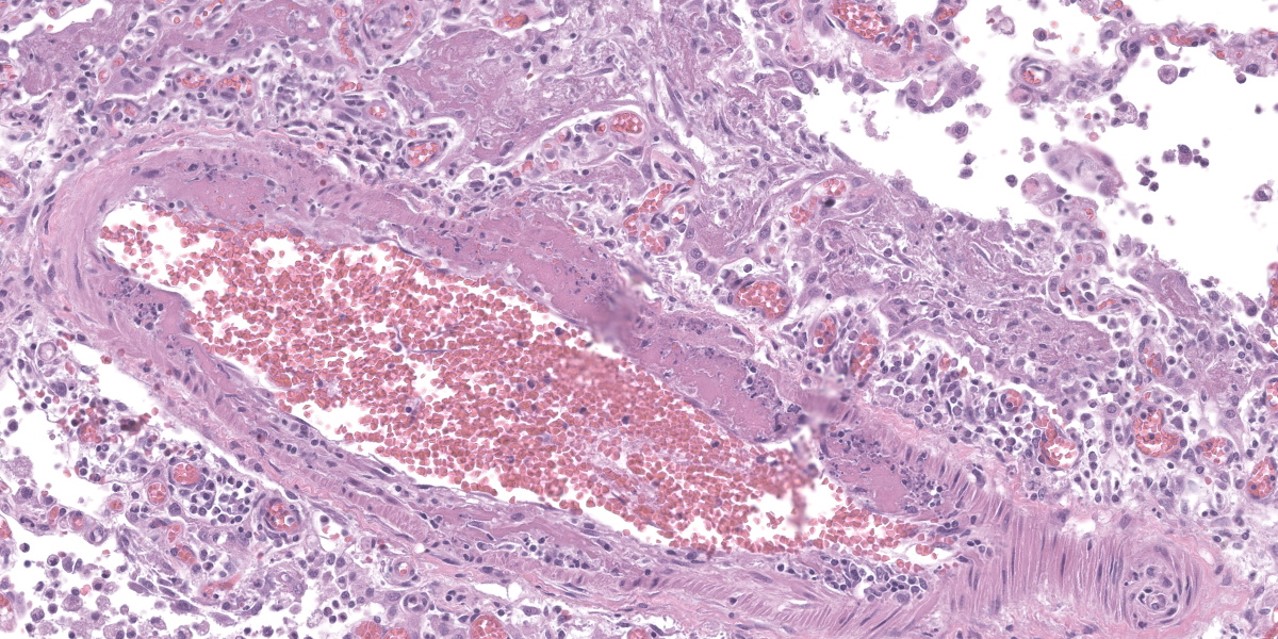
spaces contain variable amounts of fibrillar to beaded, eosinophilic material (fibrin) which often lines the alveolar septa with or without admixed cellular debris (hyaline membranes). Alveolar spaces also frequently contain finely granular, eosinophilic material (proteinaceous fluid). Within these areas as well as unaffected areas, pulmonary veins (distinguished by their location within the alveolar parenchyma) frequently exhibit remodeling, with expansion of the tunica media by plump spindle cells resulting in partial to complete occlusion of the lumen. Often associated with these remodeled veins are variably sized networks of alveolar capillaries which are moderately to markedly congested. The wall of one large pulmonary artery exhibits segmental fibrinoid necrosis.
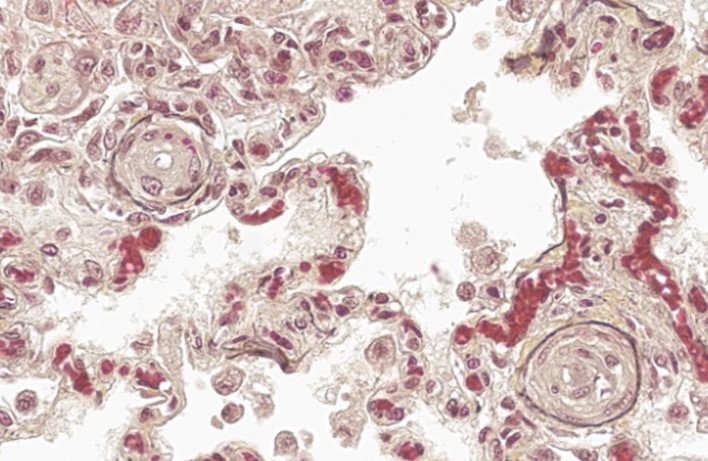
A large number of thickened vessels within the alveolar parenchyma have external, dark purple to black elastic laminae only (as seen on Verhoeff-van Gieson stain), distinguishing them as veins, in comparison to arteries which have internal and external elastic laminae. Immunohistochemical stains for smooth muscle actin (SMA) revealed SMA immopositive mesenchymal cells expanding vein walls, consistent with smooth muscle hypertrophy/hyperplasia. The cells lining the variably congested alveolar capillaries exhibit strong immunoreactive for CD31, confirming endothelial origin.
Contributor’s Morphologic Diagnoses:
- Lung: Venous medial hypertrophy, multifocal, marked, chronic, with moderate perivenous capillary congestion.
- Lung: Capillary hemangiomatosis, multifocal, marked, chronic, with minimal alveolar septal fibrosis.
- Lung: Alveolar histiocytosis, multifocal, marked, chronic, with hyaline membrane formation and moderate to marked type II pneumocyte hyperplasia.
Contributor’s Comment:
The combination of the findings of venous medial hypertrophy and alveolar capillary proliferation are consistent with pulmonary veno-occlusive disease (PVOD) and capillary hemangiomatosis (PCH). PVOD and PCH are rare forms of pulmonary hypertension (PH) which result from progressive remodeling and obstruction of pulmonary veins, and proliferation of thin-walled microvessels within alveolar septa, respectively.2,6 As the two lesions often occur concurrently, the World Health Organization (WHO) recognizes that they may belong to a spectrum of the same pulmonary vascular disease.7 In humans, the WHO recognizes five clinical groups of PH: group 1, which was recently updated to include PVOD and PCH, comprises diseases involving pulmonary arterial hypertension (PAH); group 2 represents diseases caused by left-sided heart disease; group 3 causes of PH relate to lung disease and/or hypoxia; group 4 is PH caused by pulmonary artery obstruction; and group 5 is composed of various disease with unclear and/or multifactorial mechanisms.
In a recent retrospective study of 15 dogs with PVOD and PCH, all dogs were older than 8 years of age (median 11 years).4 There was no clear sex or breed predilection, although the sample sizes were small. The underlying causes of these diseases in dogs remain unknown. In humans, patients with PVOD are predominantly young to middle-aged, and both genetic and non-genetic risk factors have been identified. Genetically, one study found that 13/13 familial and 5/20 sporadic cases of PVOD had a loss of function mutation of EIF2AK4; however, the link between this mutation and PVOD remains unclear.1 Other risk factors for PVOD in humans include chemotherapeutics, exposure to organic solvents, bone marrow or stem cell transplants, neoplasia, connective tissue disorders, autoimmune diseases, sarcoidosis, HIV infection, and pulmonary Langerhans cell histiocytosis.4 Only genetic factors have been implicated for PCH in humans.
In dogs, clinical signs involve rapidly progressive respiratory signs that include coughing, tachypnea, dyspnea, hypoxia, epistaxis, and hemoptysis.3 Diagnostically, thoracic radiographs should demonstrate an interstitial or alveolar pattern with enlargement of the pulmonary arteries, without evidence of cardiomegaly.4
Gross lesions in dogs with PVOD typically involve diffusely edematous and firm to ‘meaty’ lungs with widespread areas of hemorrhage or congestion.3 In a study of 11 dogs, there was no gross evidence of underlying cardiac disease and 2 dogs demonstrated compensatory hypertrophy of the right ventricle.8
Histologically, the hallmark feature of PVOD is extensive, patchy remodeling of small- to medium-sized pulmonary veins. This remodeling involves expansion of the vessel wall with plump spindle cells and collagen, resulting in narrowed or occluded venous lumina. As a result of this luminal occlusion, there is upstream congestion of adjacent alveolar capillaries and in some cases pulmonary arterial remodeling. PCH, which in humans is concomitantly present in 75% of PVOD cases, is characterized by proliferation of alveolar capillaries, which can extend into pulmonary arteries and veins. Additionally, there are increased numbers of alveolar macrophages which frequently contain cytoplasmic hemosiderin. In some dogs there may be small accumulations of fibrin as well as hyaline membrane formation.
Contributing Institution:
Veterinary Diagnostic Laboratory
University of Minnesota
St. Paul, MN.
https://vdl.umn.edu/
JPC Diagnosis:
Lung: Venous intimal fibrosis and smooth muscle hyperplasia and stenosis, multifocal to coalescing, severe, with septal endothelial hypertrophy (capillary hemangiomatosis), segmental alveolar capillary engorgement, alveolar histiocytosis and siderosis, and arterial medial hypertrophy.
JPC Comment:
As the contributor notes, PVOD and PCH are rare causes of pulmonary hypertension in humans and are exceedingly rare and only recently described in dogs. This case provides an excellent example of the characteristic histologic features of these conditions and the contributor provides an excellent summary of the current state of knowledge surrounding this unusual entity.
Pulmonary hypertension can be caused by four broad mechanisms: increased pulmonary blood flow, increased pulmonary vascular resistance, increased pulmonary venous pressure, or some combination of these factors.5 The American College of Veterinary Internal Medicine has recently published a proposed classification scheme that categorizes pulmonary hypertension based roughly on these broad casual mechanisms.5 The classification scheme includes many well-known causes of pulmonary hypertension including left-sided heart disease, pulmonary disease, hypoxia, congenital left-to-right cardiovascular shunts, and Dirofilaria immitis or Angiostrongylus vasorum infection.5 Under this rubric, PVOD and PCH are nested in their own subcategory under the pulmonary arterial hypertensive diseases, a categorization that may be initially confusing since the vascular lesions of PVOD preferentially affect the pulmonary veins; however, PVOD is classified as a distinct cause of pulmonary venous hypertension whose clinical expression is severe pulmonary arterial hypertension.8
The placement of PVOD and PCH in their own subcategory within the proposed classification scheme is neither merely descriptive nor academic, but instead highlights a key difference in the response of PVOD and PCH patients to typical pulmonary arterial hypertension treatments. The treatment mainstays for these conditions include the use of type-5 phosphodiesterase (PDE5) inhibitors, the most well-known of which are sildenafil and tadalafil. Human PVOD and PCH patients treated with PDE5 inhibitors can develop acute, massive, and fatal pulmonary edema due to the inability of occluded vasculature to accomodate the increased blood flow caused by these potent vasodilators.2 Though this effect has not been definitely proven in dogs, the consensus statement extrapolates from human literature and recommends administering PDE5 inhibitors only in hospital where patients can be carefully monitored for the development of pulmonary edema.5
This week’s conference moderator was Dr. Kurt Williams, Director of the Oregon Veterinary Diagnostic Laboratory and pulmonary system enthusiast. Dr. Williams waxed poetic at the start of the conference about the ability of a slide to tell a story about pathogenesis. He encouraged conference participants to spend time understanding that story and then to structure slide descriptions and interpretations to convey it accurately to others.
In this case, the story being told centers on the vasculature, so an accurate description should describe the vessels in full, to include which branches of the vascular tree and what caliber of vessels are being affected. In cases such as PVOD, where occlusive changes can make it difficult to distinguish vein from artery, a Verhoeff-van Gieson stain, which highlights vascular elastic lamina, can be helpful in characterizing the vessels; veins will have only an external elastic lamina, while arteries will have both internal and external elastic laminae. In the absence of special stains, anatomy can be useful, as arteries are typically adjacent to airways, while veins tend to branch out more diffusely into the parenchyma.
Dr. Williams noted that the hemosiderin-laden macrophages, or “heart failure cells,” associated with PVOD typically have an abundance of hemosiderin far in excess of that normally found in garden-variety heart failure. Conference participants remarked on the generally unimpressive nature of these cells in this case, while Dr. Bruce Williams cautioned against the use of this term in communications meant for clinicians, as the term could cause the inappropriate administration of positive ionotropes which are contraindicated and potentially fatal in PVOD patients.
As the contributor notes (and this case exemplifies), capillary hemangiomatosis typically accompanies PVOD. Dr. Williams noted that the capillaries in capillary hemangiomatosis are not simply congested but also larger than normal. For this reason, Dr. Williams prefers the more dramatic term, “capillary engorgement” to “capillary congestion.” Finally, Dr. Williams noted several vessels that contained foci of intimal thickening with longitudinally-oriented blood vessels embedded within. This change is characteristic of hypoxic injury in the lung and can be seen in other contexts, such as in high mountain/brisket disease in cattle.
References:
- Eyries M, Montani D, Girerd B, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet. 2014;46(1):65-9.
- Guzman S, Khan, MS, Chodakiewitz, et al. Pulmonary capillary hemangiomatosis: a lesson learned. Autops Case Rep. 2019;9(3):e2019111.
- Maxie MG, Jubb KVF, Kennedy PC, Palmer N. Jubb, Kennedy, and Palmer's Pathology of Domestic Animals. Elsevier Saunders; 2007.
- Reinero CR, Jutkowitz LA, Nelson N, et al. Clinical features of canine pulmonary veno-occlusive disease and pulmonary capillary hemangiomatosis. J Vet Intern Med. 2019;33(1):114 –
- Reinero C, Visser LC, Kellihan HB, et al. ACVIM consensus statement guidelines for the diagnosis, classification, treatment, and monitoring of pulmonary hypertension in dogs. J Vet Intern Med. 2020;34(2);549-573.
- Siddiqui NA, Charoenpong P. Pulmonary Veno-Occlusive Disease. StatPearls Publishing; 2023.
- Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53 (1):1801913.
- Williams K, Andrie K, Cartoceti A, et al. Pulmonary veno-occlusive disease: a newly recognized cause of severe pulmonary hypertension in dogs. Vet Pathol. 2016;53(4):813-22.