CASE 2: Case 1 GR (4153953-00)
Signalment:
Age: 35-42 days; Gender: 2 females and 1 male; Breed: Yorkshire; Scientific name: Sus Scrofa Domesticus; Species: Domestic Pig
History:
The present cases (three piglets) were the offspring of a sow that had been born in a litter where several littermates had died with renal lesions. This sow was experimentally artificially inseminated with semen from a boar that previously had produced litters where several piglets had died with renal lesions before weaning.
Gross Pathology:
The carcasses of these piglets were conspicuously anemic. The myocardium of the left heart ventricle was thickened due to hypertrophy. The stomach contained some coagulated blood, and erosions and hemorrhagic ulcerations were observed in the cutaneous mucosal lining of the pars oesophagea.
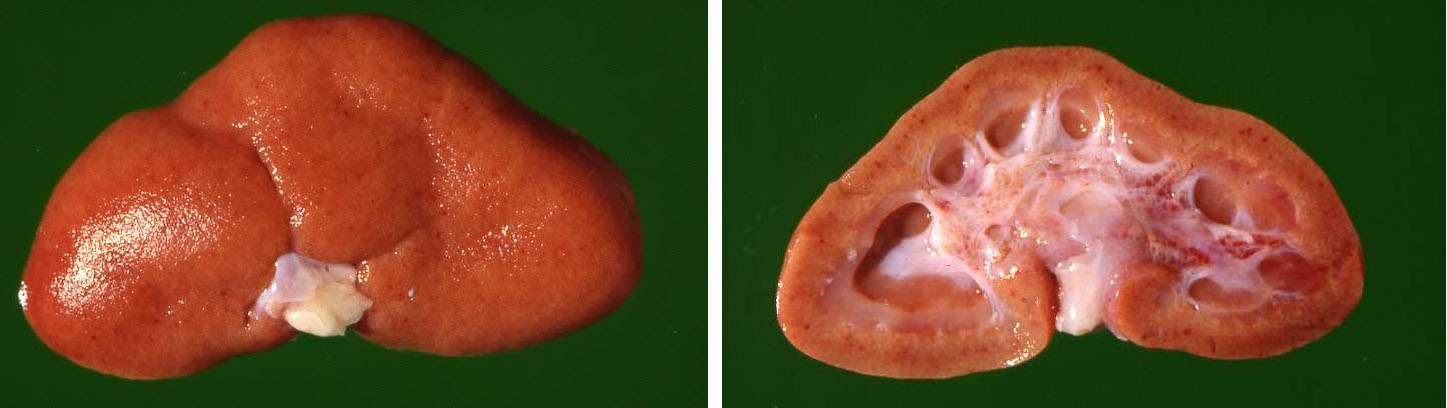
In the retroperitoneal abdomen there was a well-developed perirenal edema. The renal capsule could be easily removed. The kidneys were enlarged and somewhat pale yellow-brownish discolored with a finely granular surface containing numerous diffusely distributed cortical petechial hemorrhages. The cortical surfaces had several linear shallow indentations radiating from the renal hilum out to the renal lateral border. At outside inspection the kidneys appeared somewhat "collapsed" flattened at the hilum. After sagittal section of the kidneys the renal papillae on the cut surface appeared atrophic giving the impression of dilated calyces mimicking hydronephrotic change.
Laboratory results:
Median serum urea concentration: 44.9 mmol/L; Median serum creatinine concentration: 848 µmol/L; Microbiology (kidney, spleen and blood): No bacterial growth; Median plasma C3 concentration: 4,5 % i.e. hypocomplementemic; Median plasma Terminal Complement Complex (TCC; "MAC") concentration 13,6 Arbitrary Units (AU)/mL as compared to median plasma TCC concentration in healthy age-matched piglets of 1.8 AU/mL.
Microscopic description:
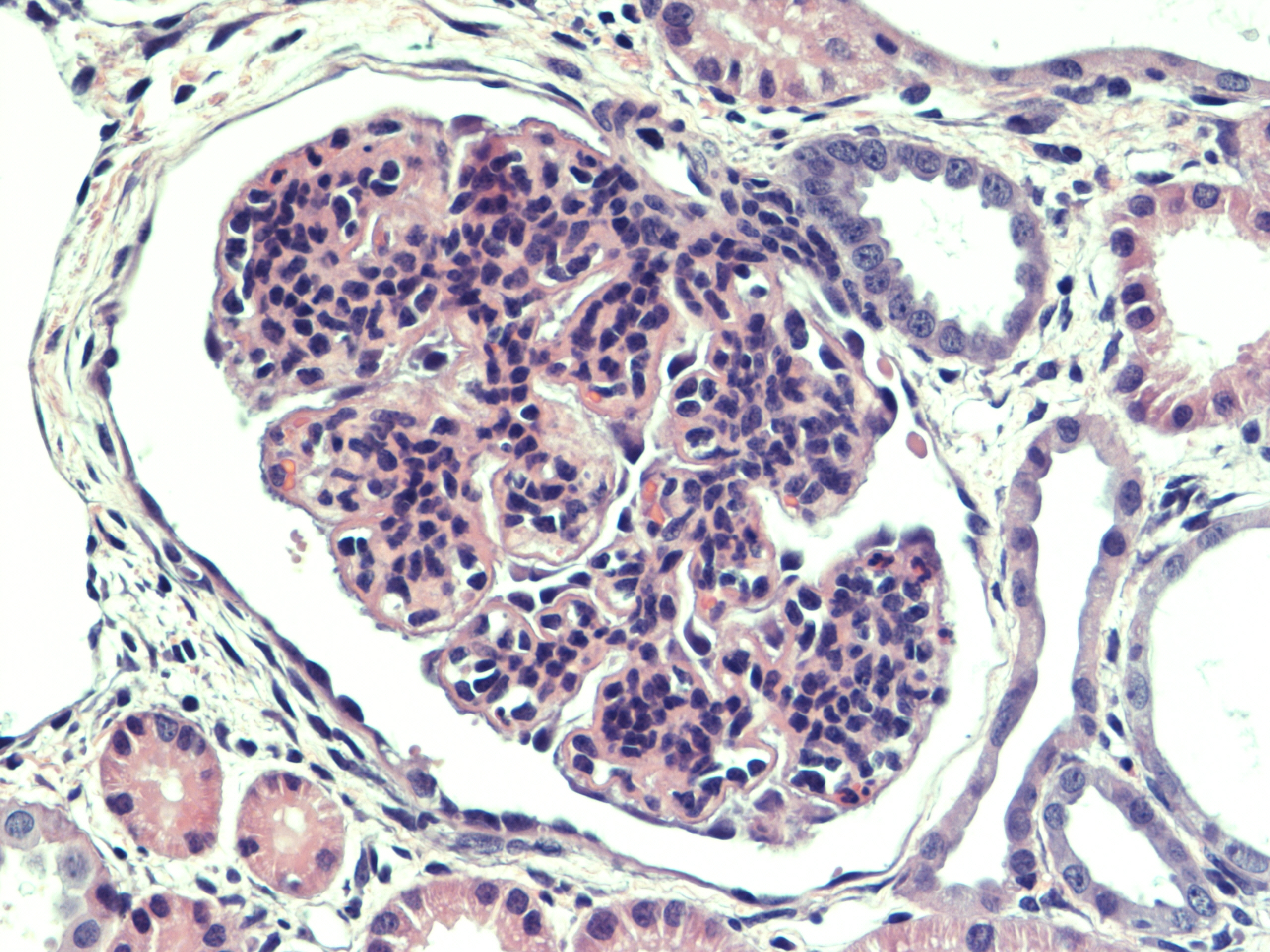
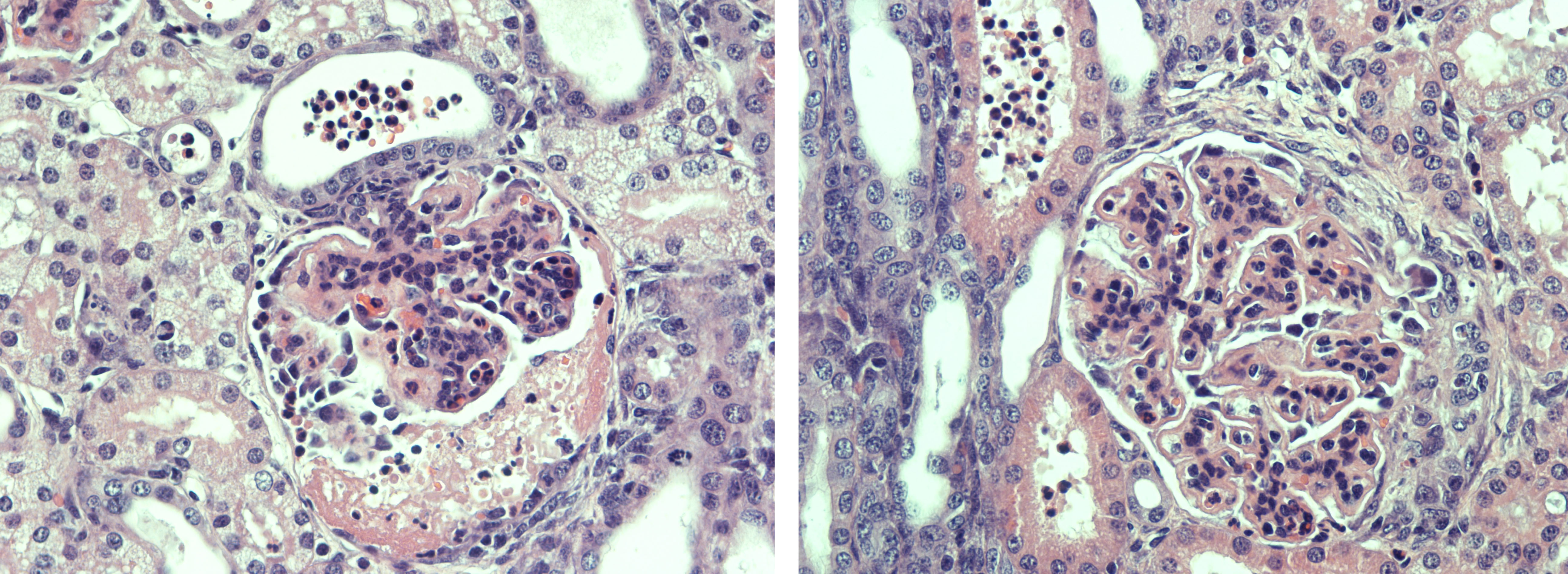
Renal tissue: All glomeruli had a principal glomerular lesion that was characterized by a combination of diffuse glomerular capillary wall thickening and pronounced mesangial cell hyperplasia. This glomerular lesion gave the glomeruli an accentuated lobulation with loss of glomerular capillary patency. Several glomeruli revealed more exudative changes with evidence of glomerular capillary infiltration of neutrophilic granulocytes (PMN-N) and fibrin exudation with a variable number of PMN-Ns to the urinary spaces (within the Bowman capsules) with a varying degree of fibrinocellular crescent formation, corresponding to a rapidly progressive glomerulonephritis (RPGN).
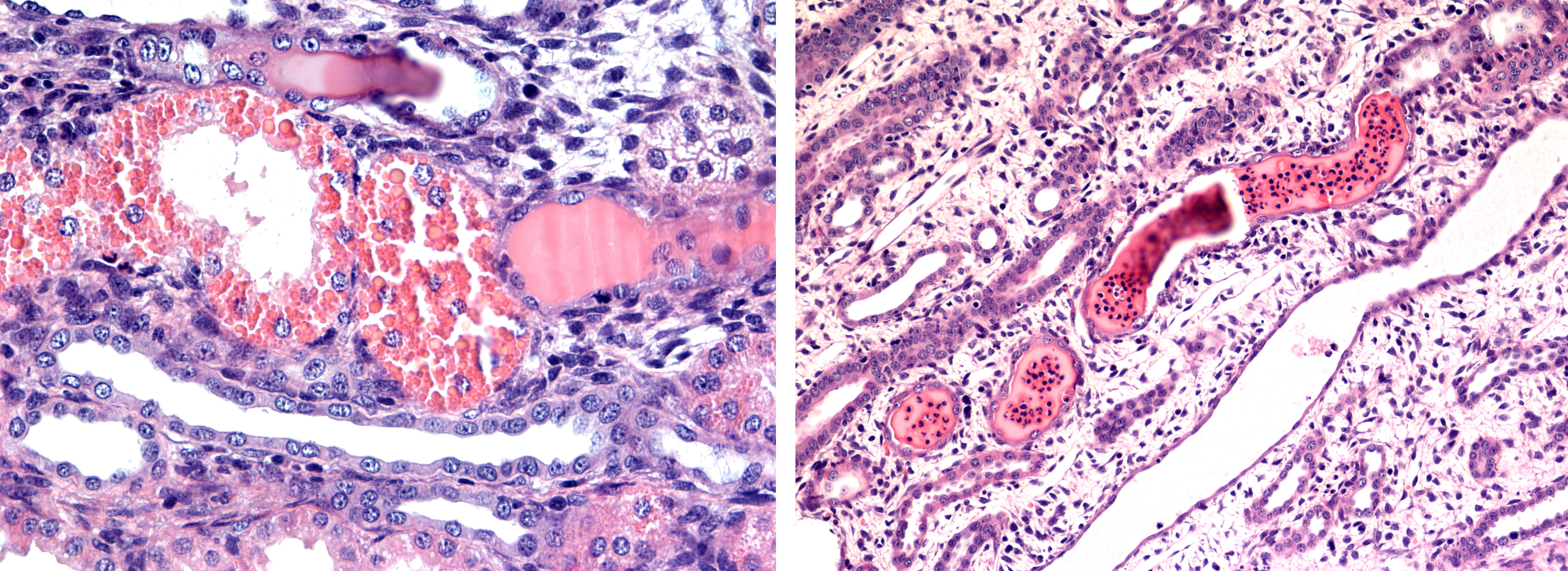
The proximal tubular epithelium revealed degenerative changes with conspicuous evidence of massive hyaline droplet degeneration. A vast number of distal tubular segments and collecting ducts contained cylinders of proteinaceous fluid with a variable content of PMN-N´s. The renal cortical interstitium was moderately expanded by collagen, and the medullary interstitium was conspicuously expanded by fibrocytes/fibroblasts. In the papillary medullary tissue, there was a scarcity of collecting ducts and other tubular elements such as the loops of Henley.
Immunohistochemistry / Immunofluorescence
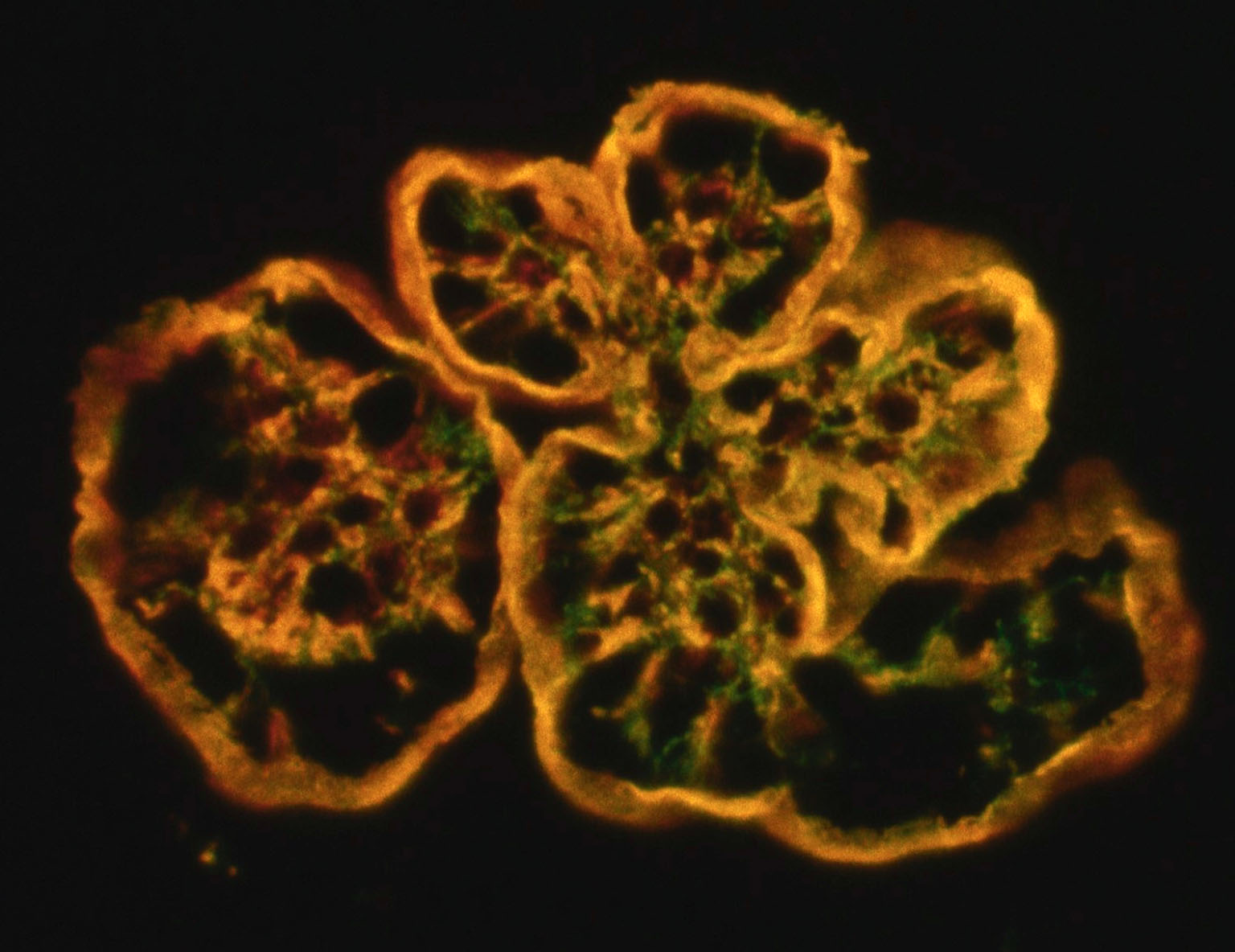
Immunohistochemistry and immunofluorescence studies of the glomeruli were consistently negative for immune-complex depositions. Indirect immunofluorescence on frozen kidney sections for glomerular porcine C3 (pAb SwineC3) was strongly positive along the glomerular capillary walls and in the expanded mesangium, and mAb HumanTCC ("MAC") was consistently strongly positive mainly along the thickened glomerular capillary walls. Double labeling showed colocalized staining (yellow) along capillary walls, while C3 deposition dominated in the mesangium. Green: FITC (fluorescein isothiocyanate. Red: TRITC (tetramethylrhodamine).
Transmission electron microscopy (TEM):
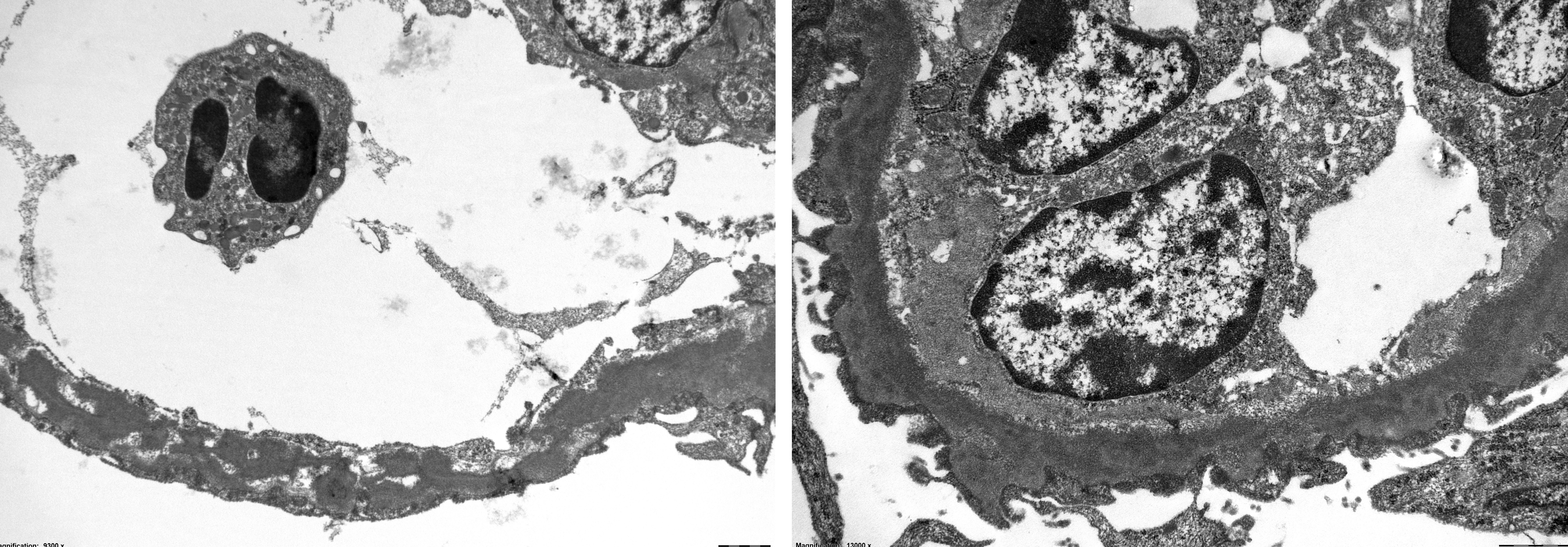
The ultrastructural appearance of the GBM revealed a conspicuously thickened lamina densa consisting of a homogenous electron dense osmiophilic material. The thickness of the GBMs were measured to 1000-1250 nm as compared to the normal GBM thickness of about 150 ? 200 nm. The glomerular visceral epithelium (the podocytes) revealed general loss of foot processes and showed a general attenuation along the outside of the thickened electron dense GBM.
Contributor's morphologic diagnosis:
Porcine Membranoproliferative Glomerulonephritis type II; Porcine C3 glomerulopathy, aka (Porcine) Dense Deposit Disease (DDD)
Contributor's comment:
Glomerular diseases represent important causes of renal failure because they cause loss of glomerular function and impair blood flow within peritubular capillaries downstream of the glomeruli, affecting the entire nephrons. Pro-inflammatory cytokines, chemokines, and growth factors produced by glomerular inflammatory reactions and transported further with the ultra-filtrate and the peritubular capillary blood may also affect and cause activation of the epithelium along the entire nephron as well as the renal interstitium, causing tubular degeneration and atrophy, and interstitial fibrosis. This adds to the devastating effects of increased protein-concentration in the pre-urine.
Glomerular diseases are termed primary when the kidneys are the only or predominant organs affected as opposed to secondary glomerular disorders caused by systemic diseases, e.g. systemic lupus erythematosus (SLE), metabolic diseases, hypertension, or amyloidosis. Thus, glomerular diseases may be caused by both non-inflammatory and inflammatory reactions. However, both the clinical manifestations and glomerular histologic changes in primary and secondary forms can be similar. In cases of glomerular disease without any obvious inflammatory component, these conditions are termed glomerulopathies as opposed to more obvious inflammatory diseases which may be termed glomerulonephritides.
Primary glomerulopathies (-nephritides) are most often caused by immune-complex depositions within in the glomeruli, either as a result of deposition of circulating immune-complexes or immune-complexes caused by antibodies reacting in situ within the glomeruli, either by binding to insoluble fixed (intrinsic) glomerular antigens or to molecules planted within the glomerulus.
The antigens responsible for the immune-complex depositions are frequently antigens of infectious microorganisms but may also be self-antigens in the case of auto-immune reactions as e.g. in SLE or in neoplasia.
Primary glomerulopathies may also be caused by hereditary disease conditions as e.g. in hereditary complement factor 3 deficiency, Alport disease ("hereditary nephritis"; mutations of collagen IV), or collagen type 3 glomerulopathy caused by a hitherto unknown mutation.
The morphologic glomerular lesions in animal immune-mediated glomerulonephritides may vary, but hypercellularity (proliferation of resident glomerular cells, leukocytic infiltration, glomerular crescent formation), GBM thickening observed as a uniform, diffuse global thickening of the glomerular capillary walls with light-microscopy, and hyalinosis and sclerosis may be observed. Both membranous glomerulopathy and membranoproliferative glomerulonephritis (MPGN) are frequently diagnosed in animal species.
The MPGN occurring in animals are lesions similar to the human MPGN type I characterized by occurrence of sub-endothelial and para-mesangial glomerular immune-complexes evident as multifocal glomerular deposits of electron-dense material interposed between the glomerular basement membrane and the glomerular capillary endothelium and in the para-mesangial regions as observed with transmission electron microscopy (TEM).1,2 These immune complexes trigger classical pathway complement activation and thus cause the inflammatory reaction.
Until 1993 another type of MPGN described in humans, MPGN type II with intramembranous dense deposits - (Dense Deposit Disease; DDD), had never been observed in any animal species, when it was discovered as the cause of disease and death in Norwegian Yorkshire piglets.6 Since then, this type of glomerulonephritis has by consensus been re-named C3 glomerulopathy, denoting a glomerular lesion characterized by C3-accumulation with absent or scanty immunoglobulin deposition.10,12
This disease condition in the Norwegian Yorkshire piglets was proven to be an autosomal recessive transmitted disease caused by an inherited deficiency of the fluid phase complement regulatory protein complement factor H (FH). The FH deficiency caused spontaneous unrestricted alternative complement activation when plasma was exposed to the glomerular basement membrane that is not protected by cell membrane-bound other complement inhibitory proteins. This porcine glomerular disease model was the first demonstration of the relationship between development of glomerular DDD and unrestricted glomerular alternative complement activation.5
In the project studying this inherited disease it was produced in all 317 piglets in a total of 28 litters, using artificial insemination of both frozen and fresh semen to sows associated to the disease condition by being littermates of piglets that had died from this disease. During this study the glomerulonephritic piglets were detected by having abnormally low plasma concentrations of Complement factor 3 (C3) and abnormally high plasma concentrations of fluid phase TCC ("MAC"). 88 (27.8 %) of the produced piglets were diagnosed as FH-deficient and developed all - without exceptions ? lethal Porcine Dense Deposit Disease (previously named Porcine Membranoproliferative Glomerulonephritis type II).
As heterozygote carriers of the genetic mutations revealed about half-normal plasma concentration of C3, we were able to detect all carriers within the Norwegian breed herds for the Norwegian Yorkshire breed. All diagnosed carriers were removed from the breeding program, and thus this disease was eradicated from the Norwegian Yorkshire pig population and has not been observed to re-emerge.3-9
Contributing Institution:
Faculty of Veterinary Medicine
Oslo, NORWAY
JPC diagnosis:
Kidney: Glomerulonephritis, proliferative and crescentic, diffuse, severe with tubular degeneration, necrosis, and regeneration, protein and cellular casts, and edema.
JPC comment:
The contributor provides an excellent review of this disease while highlighting the different subtypes of membranoproliferative glomerulonephritis (MPGN). There has been reorganization of, and recently proposed categorizations within, human MPGN. Type I and III MPGN is still related to immune complex deposition and is stratified by polyclonal or monoclonal antibody deposition. However, now three entities are grouped within a new category called C3 glomerulopathy (C3 GP), all requiring dysfunction of the alternative pathway of complement activation: Dense Deposit Disease (DDD), C3 glomerulonephritis (C3 GN), and complement factor H-related protein 5 glomerulopathy (CFHR5 GP). In order to arrive at a diagnosis of one of these diseases, a combination of light microscopy, immunofluorescence/immunohistochemistry and TEM, laboratory complement findings, and clinical history and data are required. Unfortunately, it is rare that the veterinary pathologist has access to all these data.11
The alternative pathway of complement activation relies on neither immune complexes nor bacterial sugars and is constantly in a moderate state of activation. Factor B binds to C3b and factor D cleaves the complex to form the active C3bBb that has C3 convertase activity. Regulatory mechanisms become important it a process that is constantly in action, and factors H and I play critical roles. Factor H inhibits the formation of C3 convertase, while factor I inhibits C3b by proteolysis. Deficiency of factor H results in severe secondary depletion of intact C3, while deficiency of factor I results in accumulation of C3b in plasma. As described by the contributor, Dense Deposit Disease in pigs is specifically the result of complete factor H deficiency. Murine models of this disease have successfully been created, with a targeted gene deletion of exon 3 of murine factor H gene within embryonic stem cells. The mouse model is a good representation of Dense Deposit Disease, across humans and pigs.13
Conference discussion centered briefly on the glomerular crescents in this section. When confronted with glomerular injury, pigs often make large, distinct glomerular crescents, composed of fibrin, neutrophils, sloughed parietal and visceral epithelial cells, and ultimately leading to scarring. The moderator also shared that in C3GN, the electron dense deposits seen in this disease have a multisegmented shape, with a resemblance to sausage links. Immune complex deposition results in more discrete aggregates of electron dense material.
References:
1. Alpers CE. The Kidney. In: Kumar V, Abbas AK, Fausto N, Aster JC, eds. Pathologic Basis of Disease. 8 ed. Philadelphia: Saunders Elsevier; 2010:905-969.
2. Cianciolo RE, Mohr, C. F. Urinary System. In: Grant Maxie M, ed. Jubb, Kennedy and Palmer's Pathology of Domestic Animals. 6 ed. St. Louis, Missouri 63043: Elsevier; 2016:376 - 464.
3. Hegasy GA, Manuelian T, Hogasen K, Jansen JH, Zipfel PF. The molecular basis for hereditary porcine membranoproliferative glomerulonephritis type II: point mutations in the factor H coding sequence block protein secretion. Am J Pathol. 2002;161: 2027-2034.
4. Hogasen K, Jansen JH, Harboe M. Eradication of porcine factor H deficiency in Norway. Vet Rec. 1997;140: 392-395.
5. Hogasen K, Jansen JH, Mollnes TE, Hovdenes J, Harboe M. Hereditary porcine membranoproliferative glomerulonephritis type II is caused by factor H deficiency. J Clin Invest. 1995;95: 1054-1061.
6. Jansen JH. Porcine membranoproliferative glomerulonephritis with intramembranous dense deposits (porcine dense deposit disease). Apmis. 1993;101: 281-289.
7. Jansen JH, Hogasen K, Grondahl AM. Porcine membranoproliferative glomerulonephritis type II: an autosomal recessive deficiency of factor H. Vet Rec. 1995;137: 240-244.
8. Jansen JH, Hogasen K, Harboe M, Hovig T. In situ complement activation in porcine membranoproliferative glomerulonephritis type II. Kidney Int. 1998;53: 331-349.
9. Jansen JH, Hogasen K, Mollnes TE. Extensive complement activation in hereditary porcine membranoproliferative glomerulonephritis type II (porcine dense deposit disease). Am J Pathol. 1993;143: 1356-1365.
10. Pickering MC, D'Agati VD, Nester CM, et al. C3 glomerulopathy: consensus report. Kidney Int. 2013;84: 1079-1089.
11. Schena FP, Esposito P, Rossini M. A narrative review on C3 glomerulopathy: A rare renal disease. International Journal of Molecular Sciences. 2020;21(2):525.
12. Smith RJH, Appel GB, Blom AM, et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nature reviews Nephrology. 2019;15: 129-143.
Vernon KA, Pickering MC, Cook HT. Experimental models of membranoproliferative glomerulonephritis, including Dense Deposit Disease. Contrib. Nephrol. 2011;169:198-210.