WSC 2024 Conference 11, Case 1
Signalment:
20-month-old female spayed Domestic Shorthair cat, feline (Felis catus)
History:
Sudden death with no previously identified clinical signs. The submitting veterinarian suggested congestive heart failure, feline infectious peritonitis, or toxin exposure as possible differentials.
Gross Pathology:
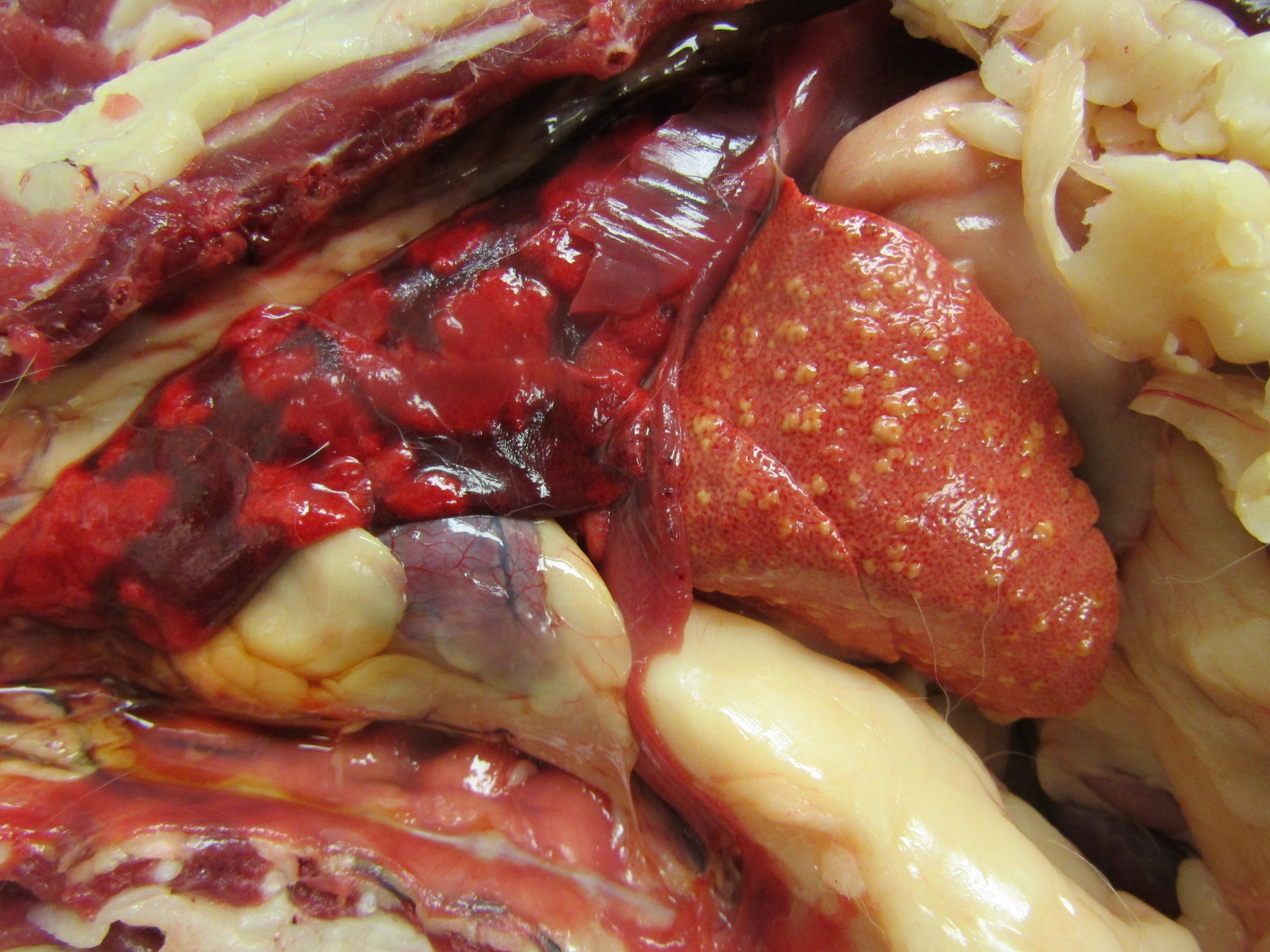
The subcutaneous and internal fat stores were mildly icteric. The liver was small and diffusely orange to brown with multifocal, slightly raised yellow nodules on the hepatic surface and throughout the parenchyma on cut section. An impression smear of the liver showed frequent spindle cells suggestive of fibroblasts or oval cells, and cells with multiple small, clear cytoplasmic vacuoles suggestive of hepatic lipidosis.
Laboratory Results:
A sample of fresh liver was submitted for mass spectrometry. A copper level of 611.2 ppm on a wet matter basis was found (normal <45ppm), which is equivalent to 2139.2 ppm on a dry matter basis.
Microscopic Description:
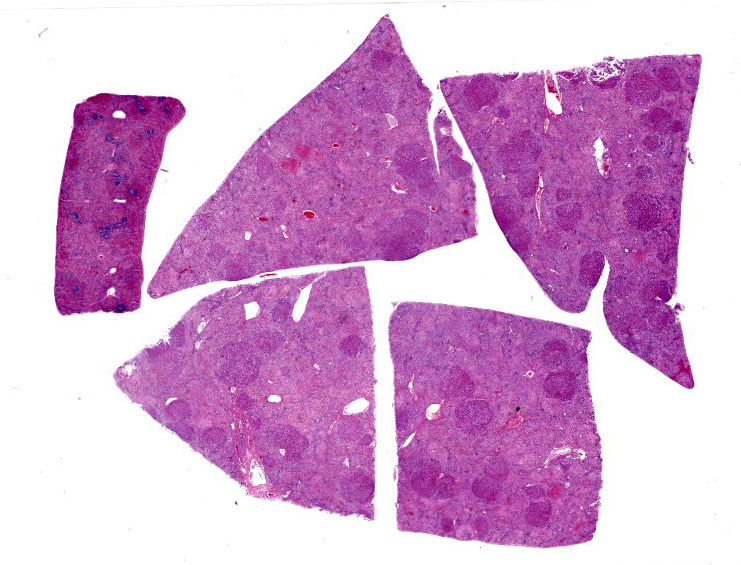
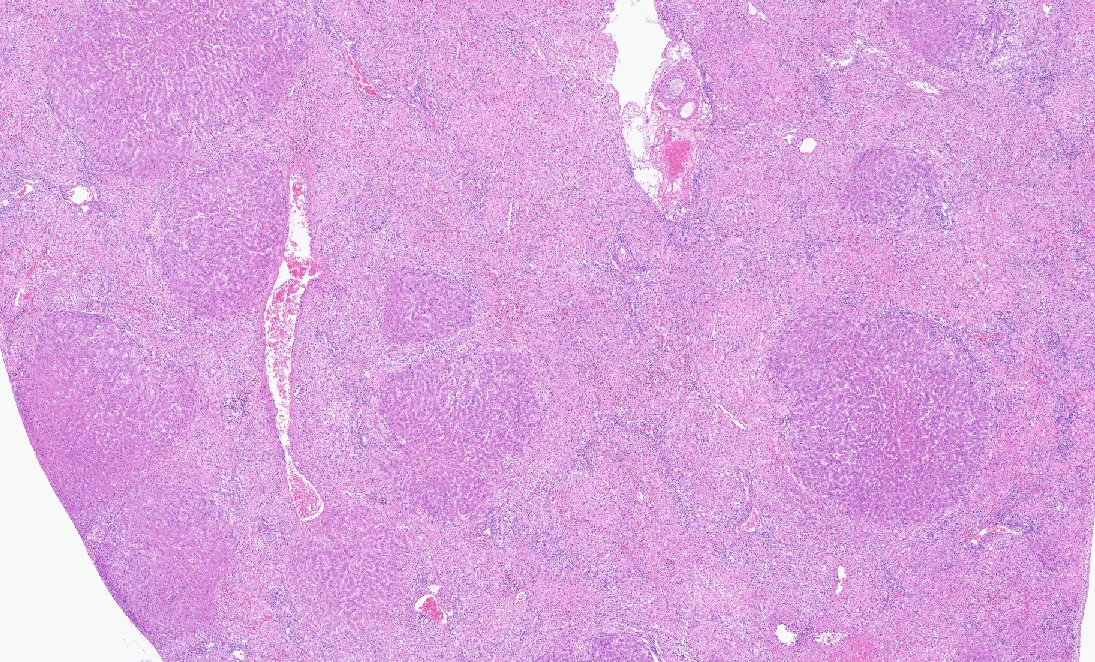
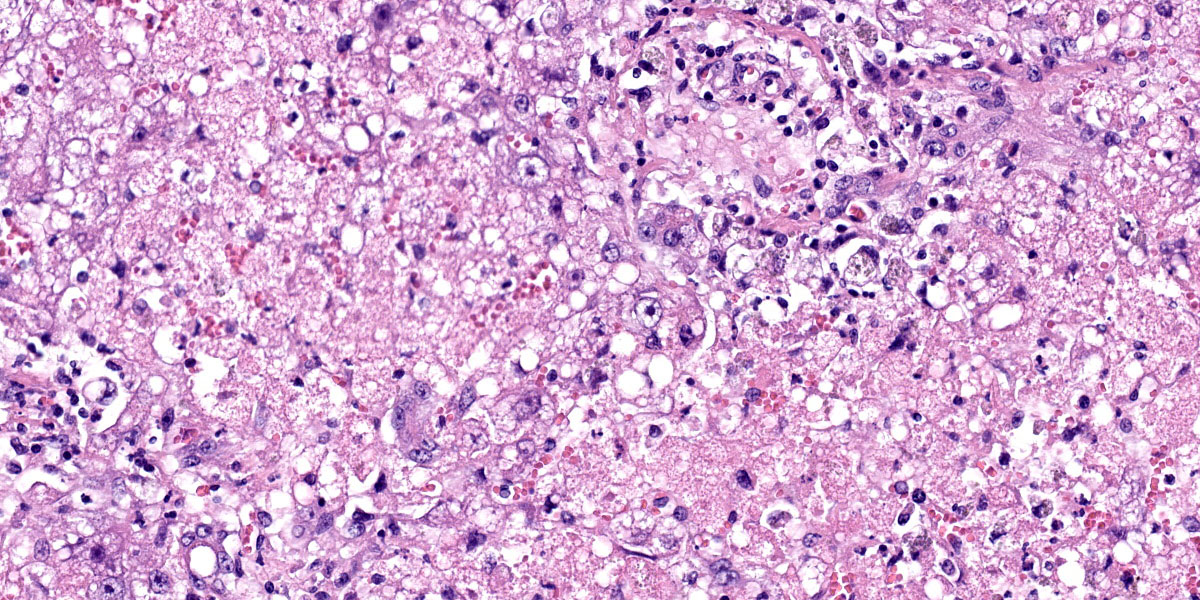
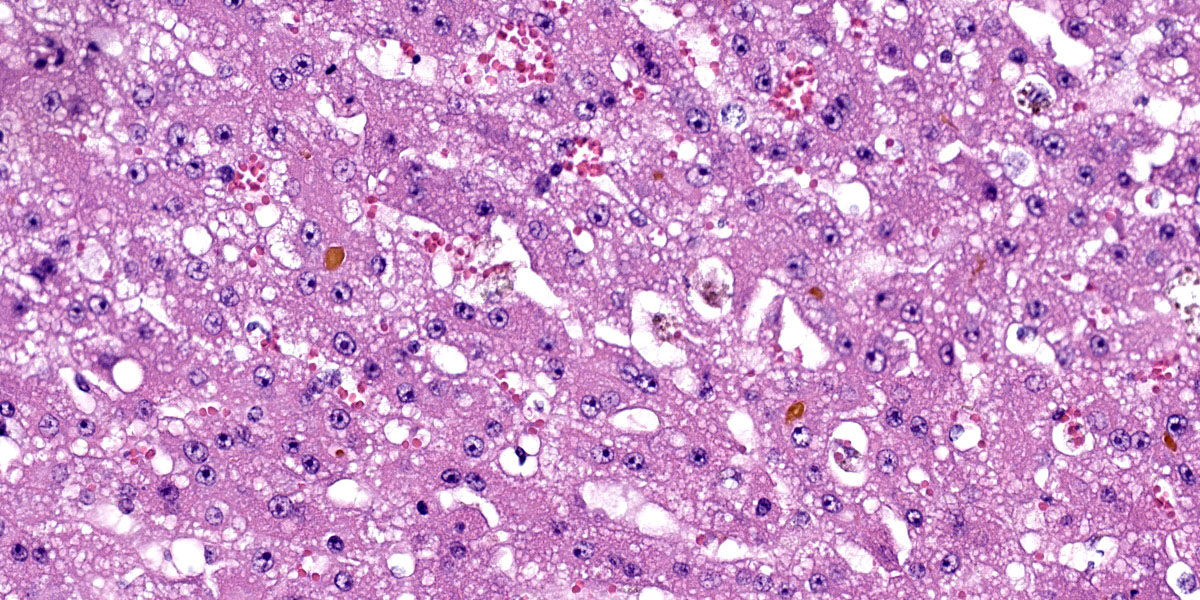
Liver: There are large coalescing areas of degeneration and necrosis of hepatocytes
affecting all zones (massive pattern) interspersed with randomly distributed, well-delimited nodules of viable hepatocytes. Portal areas and periacinar regions are close to each other (parenchymal collapse, loss of hepatocytes). Hepatocytes show marked distension of cytoplasm by small to large vacuoles (microvesicular to macrovesicular steatosis, positive Oil-red-O staining), ill-defined borders, granular eosinophilic cytoplasm, a prominent nucleus characterized by pyknosis, karyorrhexis or karyolysis. Endothelial cells are often plump (reactive) and there are reactive spindle cells (perisinusoidal cells), free pyknotic debris, and ill-defined ducts (oval cell hyperplasia). Large numbers of Kupffer cells with pale brown granules and rare red blood cells are present and similar pigment is seen in few hepatocytes.
The nodules consist of thick, disorganized hepatic plates, large hepatocytes with fine cytoplasmic vacuolation (fat, positive Oil-red-O staining), and one to two prominent nuclei. Bile canaliculi are distended by bile plugs and Kupffer cells contain large amounts of pale brown pigment (nodular regeneration). Portal areas are infiltrated by small numbers of plasma cells and lymphocytes, small amounts of collagen, and there is bile duct and oval cell hyperplasia with extension and destruction of the limiting plate. There is multifocal bridging portal-to-portal fibrosis. Terminal hepatic venules are difficult to discern. Sublobular veins are often infiltrated by small numbers of plasma cells and fat-laden macrophages. There are few foci of extramedullary hematopoiesis.
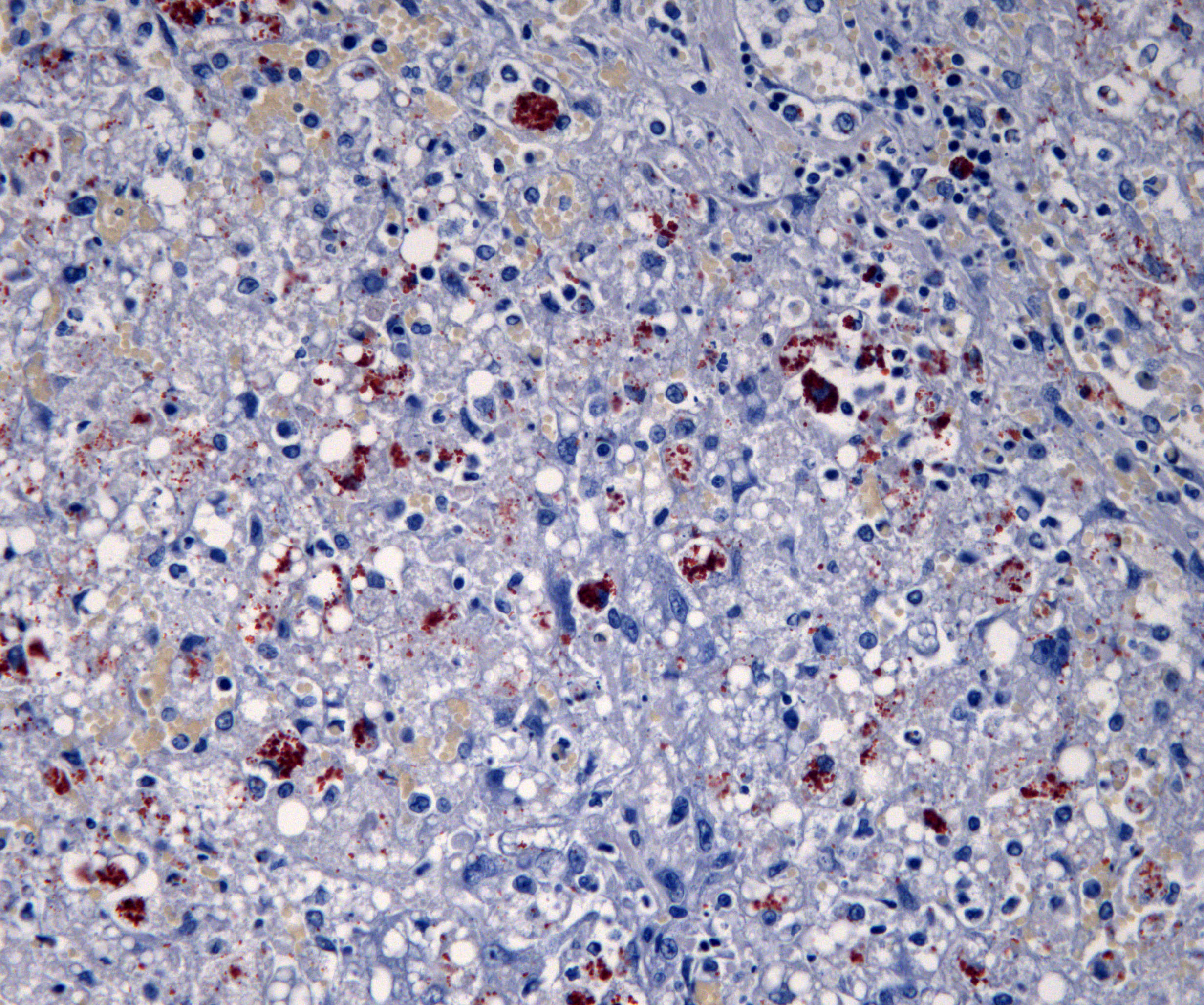
Special stains: There are large numbers of copper granules with rhodanine stain in macrophages and to a lesser degree in hepatocytes in areas of necrosis. There is a very small amount of copper in areas of regeneration. This is accompanied by large number of positive granules on Schmorl's stain (lipofuscin) and there is a moderate amount of hemosiderin in macrophages on Perl's stain. There is mild collapse and mild to moderate thickening of reticulin fibers in periacinar regions. These areas correspond to deposition of collagen with Masson’s trichrome stain. There is a small amount of collagen in periportal parenchyma.
Contributor’s Morphologic Diagnoses:
Liver: Hepatic fatty degeneration and necrosis, acute to subacute, with bile duct hyperplasia, fibrosis, nodular regeneration.
Contributor’s Comment:
Primary copper toxicity has been well-described in dogs and humans; however, it is poorly described in cats.3 The most well-known primary copper toxicity of animals occurs in Bedlington terriers, which has an autosomal recessive inheritance pattern that may be linked to a deletion in the COMMD1 genes.1 Similarly, Wilson’s disease in humans is an autosomal recessive disorder that affects the ATP7B gene, which affects secretion of copper.1 Both genes are used in copper secretion into the blood and bile.2 Without proper copper secretion, copper accumulates in hepatocytes, leading to increased cellular oxidative stress that quickly uses up intracellular glutathione.2 Ultimately, oxidative stress leads to hepatocyte necrosis.2
Previous reports in cats have been infrequent, but are often diagnosed based on being young, having histological findings consistent with copper toxicity, and no history of excessive copper intake.1,4,5 Reported histological findings in these reports include regenerative nodules, hepatic fibrosis, and hepatocytes containing brown granules that stain positively on rhodanine stain.1,5 These hepatocytes were most frequently found in the centrilobular areas.1,5 Similar rhodanine-positive granules were identified in the kidney and lung.4 These findings are similar to copper toxicity in dogs.1,5 In a study examining 104 feline liver biopsies, only 5 cases were identified with centrilobular copper.7 A similar study examining liver samples from 100 cases of hepatic disease in cats found 11 presumed cases of primary copper toxicity.5 Both of these studies suggest that primary copper toxicity is very rare.5,7
To date, the causative gene and potential inheritance pattern of primary copper toxicity in cats is unknown. One reported case of two sibling cats with primary copper toxicity showed two single-nucleotide variations in the ATP7B gene, the same gene affected in humans.1 A subsequent study identified single-nucleotide variations in ATP7B in three of four cats examined, suggesting this is the most likely cause of primary copper toxicity in cats.2
Contributing Institution:
Western College of Veterinary Medicine
University of Saskatchewan
Saskatoon, SK Canada S7N 5B4
https://wcvm.usask.ca/departments/vet-pathology.php
JPC Diagnosis:
- Liver: Hepatocellular degeneration and necrosis, massive, with marked hepatocellular lipidosis.
- Liver: Nodular hepatocellular regeneration, multifocal, moderate with mild biliary hyperplasia.
JPC Comment:
Copper is an essential heavy metal that is required for the proper function of a wide variety of enzymes, including the cytochrome oxidases (mitochondrial respiration), lysyl oxidase (collagen synthesis), tyrosinase (melanin synthesis), ceruloplasmin (iron synthesis), and antioxidant defense (superoxide dismutase).3 In excess, the multiple redox states of copper can lead to free radical production and oxidative damage to cellular components, and animals have responded to this threat by developing regulatory mechanisms to ensure excess copper is rendered inert and excreted.
Copper metabolism begins in the gut, where dietary copper is actively transported across the mucosal surface of small intestinal enterocytes. This function is performed by a specific copper transporter, Ctr1, and by the non-specific transporter, divalent metal transporter (DMT1), which also transports several trace elements (e.g., iron and zinc) with which copper competes for transport. Once in the enterocyte, copper is incorporated into enzymes or, if in excess, bound to metallothionein and stored in lysosomes to protect the cell from free copper.6 Once metallothionein is saturated, copper is excreted into the blood, where it is bound to the carrier proteins ceruloplasmin and transcuprein, or to albumin, and transported to the liver via the portal blood.6
Copper enters the hepatocyte via the same Ctr1 transporter found in the enterocytes. Once in the hepatocyte cytoplasm, chaperone proteins send copper to mitochondria and to the Golgi body, where copper is transported into the Golgi by the transporter ATP7B. ATP7B directs the incorporation of copper into ceruloplasmin for return to the systemic circulation and distribution to other tissues.6 If the ceruloplasmin/copper complex returns to the liver, it is taken up into hepatocytes and secreted into bile for excretion. If copper accumulates in excess of metabolic requirements within the hepatocytes, it is complexed with metallothionein or glutathione and stored within lysosomes as in enterocytes.6
Perturbations in any of the above processes, such as a primary metabolic defects in hepatic copper metabolism, altered hepatic biliary excretion of copper, or excess dietary copper intake, can result in hepatic copper toxicosis.3 Species susceptibility to copper toxicosis varies widely, with sheep being most prone to copper poisoning due to a lack of sufficient capacity to excrete copper into the bile and to a very small copper metallothionein binding capacity.3 When faced with a sudden dietary excess, biliary excretion can’t keep up with the influx of copper, leading to very high levels of copper deposition within the liver. When the binding capacity of metallothionein is exceeded, a sudden, generally fatal intravascular hemolytic crises can occur.3,6
The contributor provides an excellent survey of the few cases of feline copper toxicity reported in the veterinary literature which, for all their rarity, present with familiar histologic features: severe fibrosis, regenerative nodules, and excessive intracytoplasmic copper as evidenced by rhodanine staining. Copper toxicity does not, however, typically present with abundant hepatic necrosis, and the significant necrosis in the examined section provoked robust discussion before, during, and after this week’s conference.
The moderator of today’s conference was Dr. Anna Travis, Chief of Education Operations at the Joint Pathology Center, who began discussion of this case with a review of copper metabolism, as summarized above, and a review of the typical histologic changes associated with copper toxicity. Conference participants noted the small amount of fibrosis evident on Masson trichrome stain, which was surprising given the significant number of regenerative nodules.
Conference participants also noted the abundant, multifocal to coalescing hepatic necrosis, which for many was the predominate histologic feature of this case. The abundant necrosis also hampered qualitative evaluation of the rhodanine staining, which, in the remaining intact hepatocytes, appeared sparse in relation to the incredibly high copper levels measured in this patient. While participants agreed that the amount of rhodanine staining was abnormal, many felt that the amount of staining was not consistent with the dry weight of copper value obtained in the lab results. Finally, the participants felt that a second, acute insult resulted in widespread necrosis which may have obscured a more traditional pattern of feline copper toxicity.
This case was sent for post-conference consult with Dr. John Cullen, Distinguished Professor at the North Carolina State University College of Veterinary Medicine. Dr. Cullen agreed that the histologic picture in this case is not a perfect match for copper toxicity as a sole insult. Copper accumulation in reported feline cases is typically centrilobular and has a substantial inflammatory component, neither of which is apparent in this case.
Though a tidy resolution for this case remains elusive based on a complex interplay of acute and chronic pathogeneses, conference participants continue to believe that the primary lesion in this case is an acute, necrotizing insult, the cause of which is not apparent. The excessively high copper dry weight almost certainly contributed to the observed pathology in this case; however, the substantial acute necrosis is confounding, and participants preferred to separate the morphologic diagnoses into acute and chronic effects to emphasize the presumed dual pathologic processes in this fascinating case.
Getting short shrift this week was the forlorn section of spleen, located literally and figuratively on the periphery and characterized by mild to moderate chronic diffuse congestion with hemosiderin-laden macrophages.
References:
1. Asada H, Kojima M, Nagahara T, et al. Hepatic copper accumulation in a young cat with familial variations in the ATP7B gene. J Vet Intern Med. 2019;33:874–878.
2. Asada H, Chambers JK, Kojima M, et al. Variations in ATP7B in cats with primary copper-associated hepatopathy. J Feline Med Surg. 2020;22:753-759.
3. Cullen JM, Stalker MJ. Liver and Biliary System. In: Maxie GM, ed. Vol. 2, Jubb, Kennedy, and Palmer’s Pathology of Domestic Animals. Elsevier, Inc;2016: 302, 342-343.
4. Haynes JS, Wade PR. Hepatopathy associated with excessive hepatic copper in a Siamese cat. Vet Pathol. 1995; 32:427–429.
5. Hurwitz BM, Center SA, Randolph JF, et al. Presumed primary and secondary hepatic copper accumulation in cats. J Am Vet Med Assoc. 2014;244:68–77.
6. Lopez-Alonso M, Miranda M. Copper supplementation in cattle: a challenge. Animals (Basel). 2020;10(10):1890.
7. Whittemore JC, Newkirk KM, Reel DM, et al. Hepatic copper and iron accumulation and histologic findings in 104 feline liver biopsies. J Vet Diagn Invest. 2012; 24:656–661.