WSC 2023-2024. Conference 2, Case 1
Signalment:
2-year-old, female spayed Australian cattle dog, canine (Canis lupus familiaris)
History:
Unspecified clinical signs began at 11-12 months of age. An MRI performed at 21 months of age revealed brain atrophy. Clinical signs progressed to ataxia, anxiety, blindness, and aggression and the animal was euthanized at 23 months of age.
Gross Pathology:
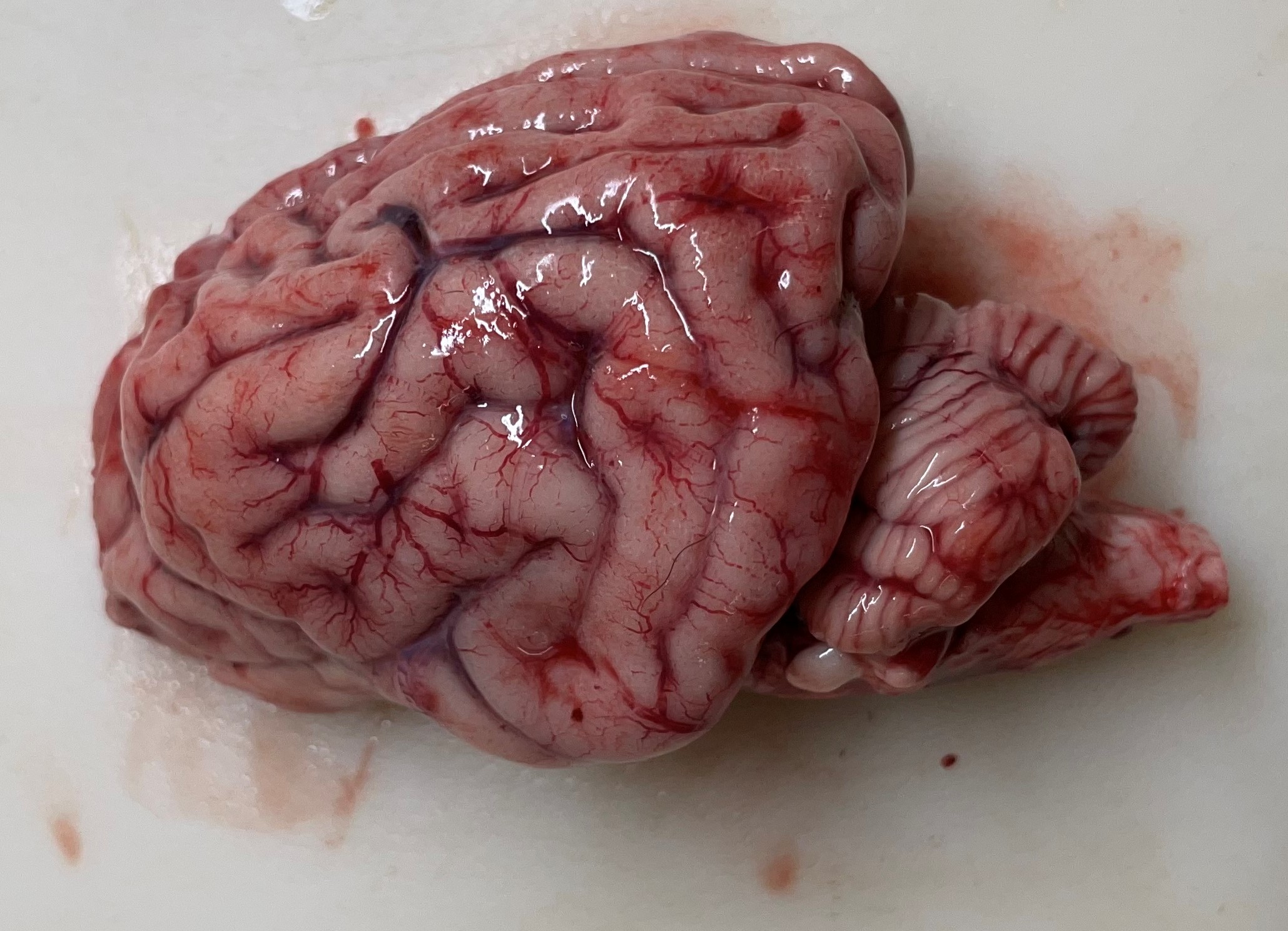
The cerebral gyri are uniformly narrow and widely separated by deep, prominent sulci. The cerebellar folia in all lobes are more deeply incised than expected for a normal brain. There are no other significant findings.
Microscopic Description:
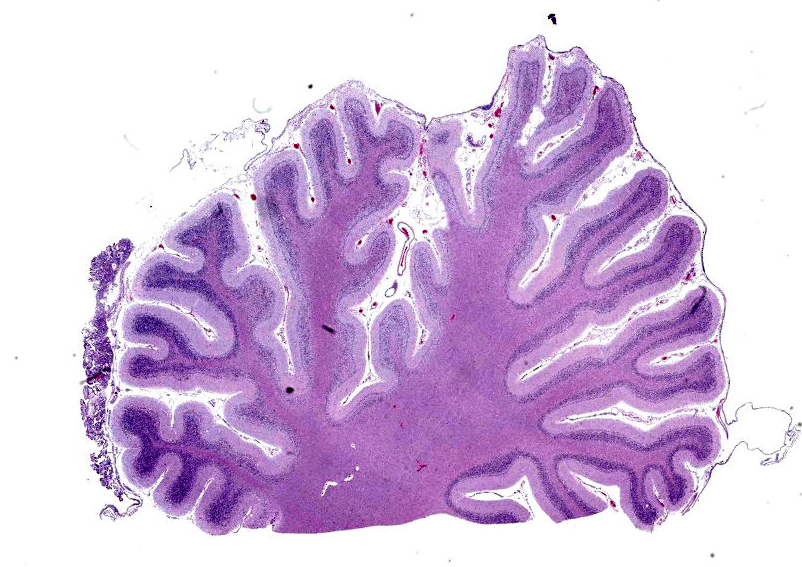
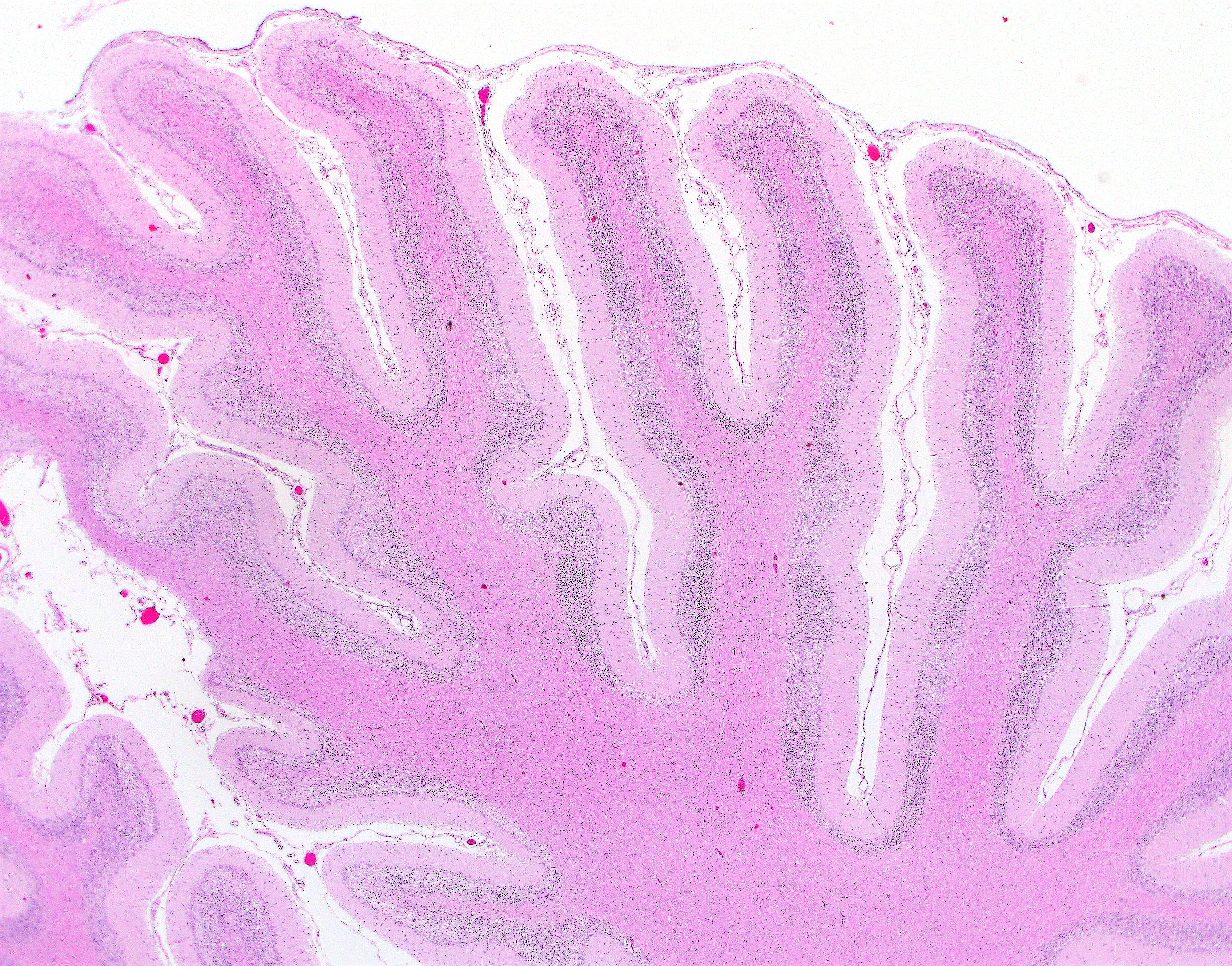
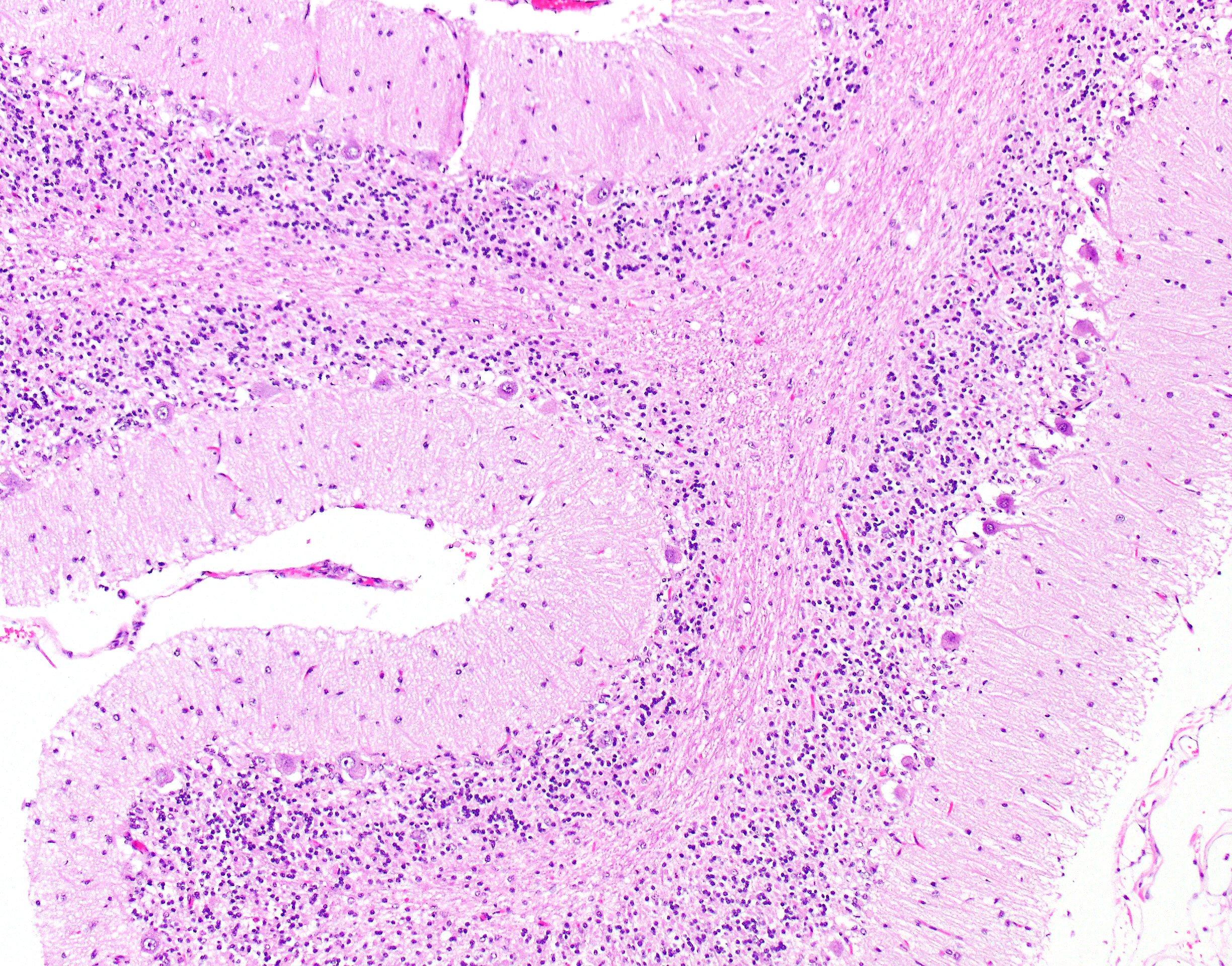
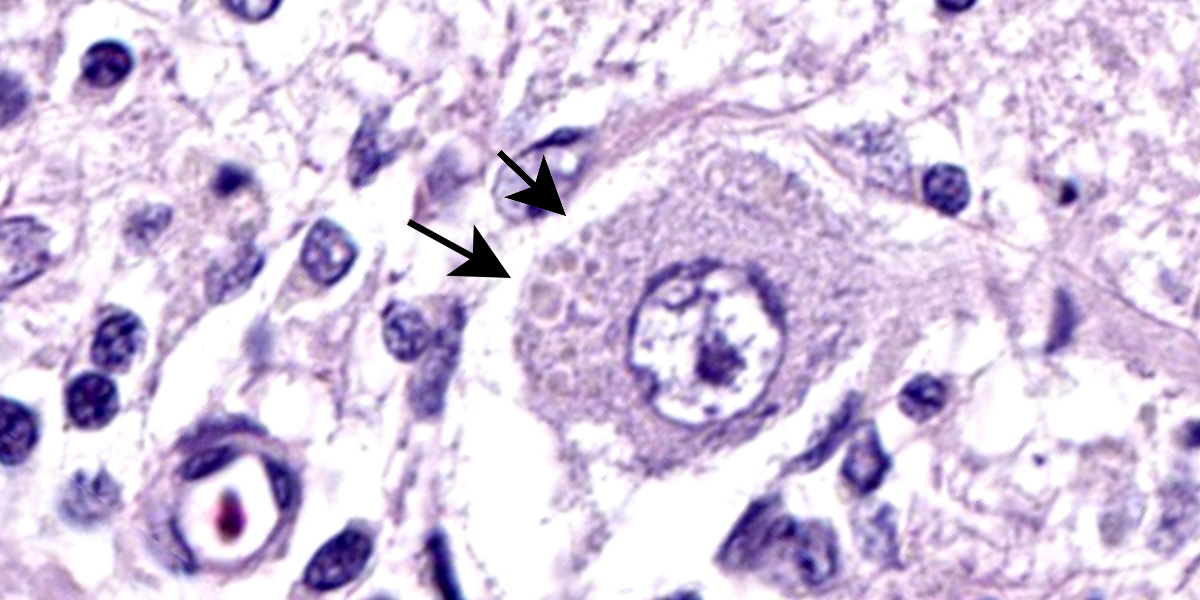
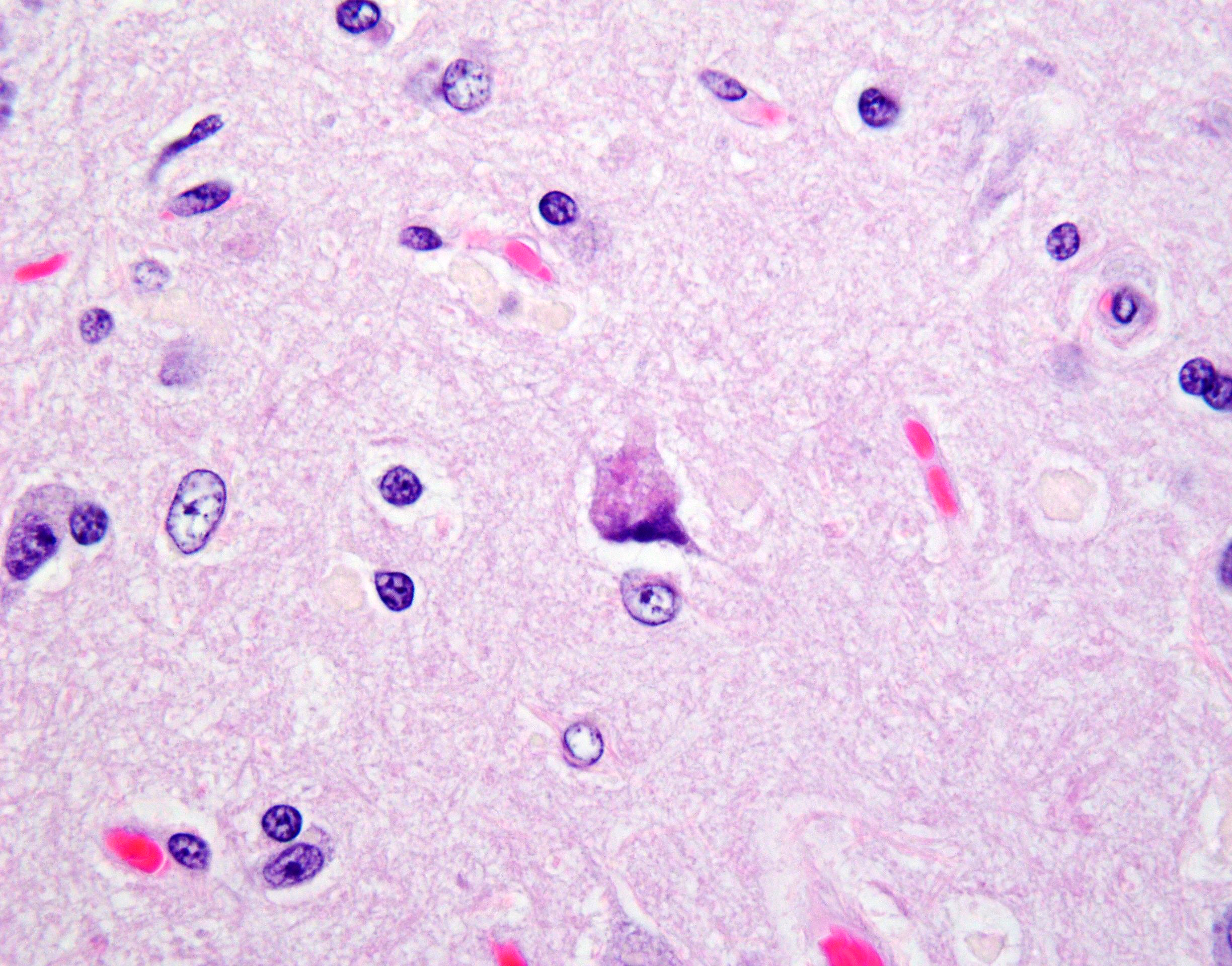
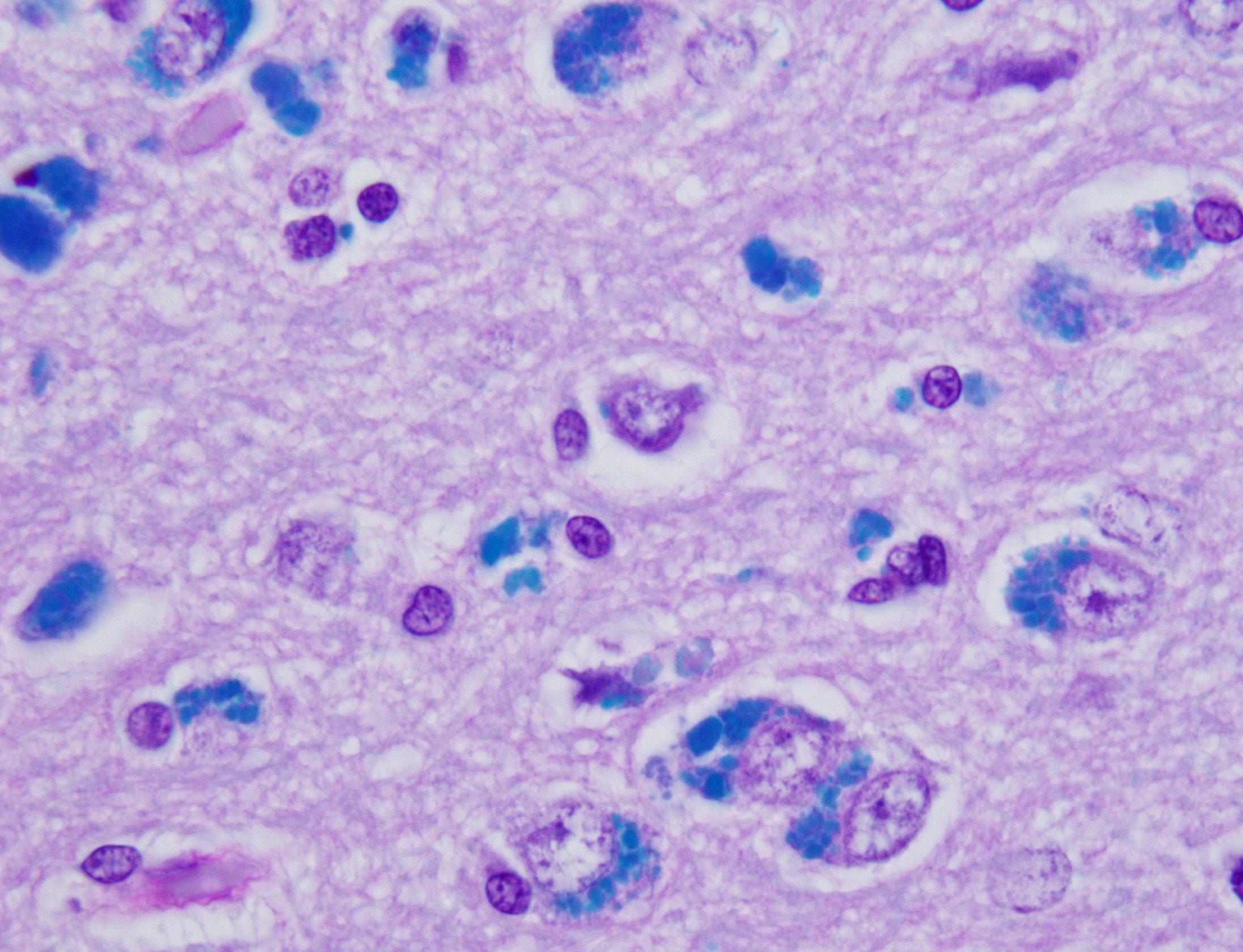
Cerebellar atrophy is manifest as slender folia that retain an anatomically normal branching pattern. This change is quantitatively similar throughout the cerebellum. At higher magnification, the molecular layer is narrow, and the internal granule layer is also thin and depleted of cell nuclei. Interestingly, the Purkinje cell layer is variably altered and the distance between cells is sometimes less than normal, possibly due to shrinkage in other layers. Neuronal cell bodies are multifocally swollen by variable amounts of cytoplasmic, granular to globoid, lightly eosinophilic to brown pigment which occasionally peripheralizes the Nissl substance or nucleus. Rare individual neurons are brightly eosinophilic and shrunken with pyknotic nuclei. Sections stained with Luxol fast blue-PAS highlighted cytoplasmic inclusion bodies consistent with lipofuscin.
Contributor’s Morphologic Diagnosis:
Brain: Cerebellar cortical atrophy with intracytoplasmic neuronal storage material consistent with lipofuscin.
Contributor’s Comment:
The neuronal ceroid lipofuscinoses (NCLs) are a group of inherited neurodegenerative diseases characterized by progressive decline following normal development. Characteristic of the group is accumulation of auto fluorescent pigment contained in lysosomes of neurons and other cell types.11
Dogs are useful models of pathogenesis for this group of diseases and can be used to test the efficacy of therapies for humans.4 There are 13 canine sequence variants in 8 canine NCL orthologs of human NCLs that produce pathology similar to human diseases and various NCLs have been discovered in 20 different dog breeds.4 The various subtypes of NCLs are referred to by the name of the particular mutated gene: “CLN,” meaning ceroid lipofuscinosis, neuronal, followed by a number.
CLN types 1-4 are diseases of late infantile or juvenile onset, compared to CLNs 5-8 that have infantile onset.11 There are 27 different mutations known in humans.11 In CLN5 disease, affected dogs start showing clinical signs at around a year of age. Similar to this dog, the pathology in other breeds and humans consists of severe, generalized, progressive cerebral and cerebellar atrophy. The defective gene product is a soluble glycoprotein that is cleaved in the endoplasmic reticulum and is transported to lysosomes and cleaved by mannose-6-phosphate.6,11 Intraneuronal storage of the subunit c of mitochondrial ATP synthase and Saponins A and D follows.11 For all forms of CLN the end product is highly fluorescent and examination of unstained sections by fluorescence microscopy is the most sensitive form of detection.4 Electron-dense ultrastructural deposits are similar but not identical between the different types of lipofuscinosis.11
This patient was an Australian cattle dog that began showing neurological signs at 11-12 months of age. MRI imaging detected diffuse brain atrophy at 21 mos and the dog was euthanized at 23 mos. CLN5 in this breed of dog is the result of a homozygous C->T transition at position 30,574,637 on chromosome 22, reflected in the transcript (CLN5:c:619C>T). This change coverts a glutamine to a termination codon (p.Gln207TER).5 An identical mutation has been found in border collies and Laborador-Beagle mix dogs.7,8,10 A second autosomal recessive mutation in the CLN5 gene (c.934_935delAG) causes disease in Golden Retrievers.2 Purebred dogs often concentrate homozygous animals, leading to affected offspring. For instance, a high mutant allele frequency (34.8%) has been found in kennels of border collie dogs in Japan, with an allele carrier frequency of 8.1%.7,8 Popular breeds may also develop more than one CLN mutation (e.g., CLN8 in Australian shepherd dogs and CLN12 in Australian cattle dogs).3,9
Increasingly, inborn errors of metabolism are being identified in mixed breed dogs in which neither parent is clinically affected. That several breeds may have a high incidence of heterozygosity and produce affected offspring may suggest that some locations are genetic hotspots in dogs. Finding a CLN5 affected Laborador-Beagle dog is presumably an example to this occurrence.10 Recent studies of large populations of dogs suggest that some recessive mutations may occur at moderate frequencies in a number of different breeds so that inborn errors of metabolism are increasingly likely in mixed breed dogs.1,12
Contributing Institution:
University of Missouri
Veterinary Medical Diagnostic Lab
https://vmdl.missouri.edu/
JPCDiagnosis:
Cerebellum: Cortical atrophy, diffuse, with neuronal ceroidosis.
JPC Comment:
The storage diseases that collectively comprise the neuronal ceroid lipofuscinoses are rather poorly named, as the storage material is neither exclusively ceroid nor lipofuscin. Instead, as the contributor notes, compounds such as subunit c of mitochondrial ATP synthase and sphingolipid activator proteins A and D may constitute the bulk of the retained material.2
Inherited neuronal ceroid lipofuscinoses have been described in a variety of cat, sheep, dog, and cattle breeds and are a heterogenous group of diseases. The various disease presentations reflect the diversity of genetic mutations that underly the disease entities, with most mutations occurring primarily in genes that code for lysosomal enzymes (ARSG, ATP13A2, CLN5, CTSD, PPT1, TPP1), endoplasmic reticulum proteins (CLN6), and endoplasmic reticulum-Golgi complex intermediate compartments (CLN8).3 All mutations result in the inappropriate storage of proteins and lipofuscin-like lipopigments in multiple organs, but the most clinically significant damage occurs in the cerebral cortex, retina, and cerebellar Purkinje cells.2 Lesions in these regions result in extensive cellular loss and atrophy with the concomitant clinical signs of dementia, blindness, and ataxia.2
|
Breed |
Mutated Gene |
|
Devon cattle |
CLN5 |
|
English Setter |
CLN8 |
|
Border Collie |
CLN5 |
|
Australian Shepherd |
CLN6 |
|
American Bulldog |
CTSD |
|
Staffordshire Terrier |
ARSG |
|
Tibetan Terrier |
ATP13A2 |
|
Dachshund |
PPT1 |
|
Miniature Dachshund |
TPP1 |
|
Borderdale sheep |
CLN5 |
|
South Hampshire sheep |
CLN6 |
|
Table 1-1. Selected breeds with associated NCL-related mutations. |
|
Disease presentation, even among species with the same gene mutation, can be subtly different. For instance, the disease presentation in Border Collies and a few other related dogs breeds are due, as the contributor discusses above, to mutation in the CLN5 gene. In these animals, there are gait and visual deficits with increasing aggression and dementia by 18-24 months of age, a case presentation that mirrors the clinical history in this case.2 The resulting blindness is central in nature as retinal lesions are usually mild. The CLN5 gene deletion typically concentrates neuronal loss in the Purkinje cell layer and the limbic system, resulting in the ataxia and behavior changes that characterize this particular NCL.2 By contrast, the mutated bovine CLN5 in Devon cattle causes blindness by 14 months of age due to severe retinal atrophy but with only mild neuronal loss within the cerebrum and cerebellum.2
While the underlying pathogeneses of the NCLs are still largely unknown, diagnosis is relatively straightforward. NCL-associated storage granules are characteristically autofluorescent under ultraviolet light, have characteristic ultrastructural lamellar profiles, are PAS- and Luxol fast blue-positive, and are weakly acid fast.2,12
Conference discussion led by this week’s moderator, MAJ Brittany Beavis, Chief of Molecular Pathology at the United States Army Medical Research Institute of Infectious Diseases, centered on the Purkinje cell layer, which appeared atrophic in some sections and relatively normal in others. Conference participants discussed how Purkinje cells can be subject to neurogenic atrophy due to loss of granule cells in addition to the direct damage caused by inappropriate retention of storage material. The variable ratio of these damaging inputs within the affected tissue could be one reason for the heterogeneity observed within the Purkinje cell layer and is illustrative of the interconnectivity among the cerebellar layers and among neural structures more generally.
References:
- Donner J, Anderson H, Davison S, et al. Frequency and distribution of 152 genetic disease variants in over 100,000 mixed breed and purebred dogs. PLoS Genet. 2018;14(4):e1007361.
- Cantile C, Youssef S. Nervous System. In: Maxie MG, ed. Jubb, Kennedy and Palmer’s Pathology of Domestic Animals. Vol 1. 6th ed. Elsevier; 2016: 290-291.
- Chalkley MD, Armien AG, Gilliam DH, et al. Characterization of neuronal ceroid-lipofuscinosis in 3 cats. Vet Pathol. 2014;51(4):796-804.
- Gilliam D, Kolicheski A, Johnson GS, et al. Golden Retriever dogs with neuronal ceroid lipofuscinosis have a two-base-pair deletion and frameshift in CLN5. Mol Genet Metab. 2015;115(2-3):101-9.
- Guo J, Johnson GS, Brown HA, et al. A CLN8 nonsense mutation in the whole genome sequence of a mixed breed dog with neuronal ceroid lipofuscinosis and Australian Shepherd ancestry. Mol Genet Metab. 2014;112(4):302-9.
- Katz ML, Rustad E, Robinson GO, et al. Canine neuronal ceroid lipofuscinoses: Promising models for preclinical testing of therapeutic interventions. Neurobiol Dis. 2017;108:277-287.
- Kolicheski A, Johnson GS, O'Brien DP, et al. Australian cattle dogs with neuronal ceroid lipofuscinosis are homozygous for a CLN5 nonsense mutation previously identified in border collies. J Vet Intern Med. 2016;30(4):1149-58.
- Mamo A, Jules F, Dumaresq-Doiron K, et al. The role of ceroid lipofuscinosis neuronal protein 5 (CLN5) in endosomal sorting. Molec Cell Biol. 2012;32:1855-1866.
- Mizukami K, Chang HS, Yabuki A, et al. Neuronal ceroid lipofuscinosis in border collie dogs in Japan: clinical and molecular epidemiological study (2000-2011). ScientificWorldJournal. 2012; 2012:383174.
- Mizukami K, Chang HS, Yabuki A, et al. Novel rapid genotyping assays for neuronal ceroid lipofuscinosis in border collie dogs and high frequency of the mutant allele in Japan. J Vet Diagn Invest. 2011;23(6):1131-1139.
- Schmutz I, Jagannathan V, Bartenschlager F, et al. ATP13A2 missense variant in Australian cattle dogs with late onset neuronal ceroid lipofuscinosis. Mol Genet Metab. 2019;127(1):95-106.
- Vandevelde RJH, Oevermann A. Veterinary Neuropathology: Essentials of Theory and Practice. 1st ed. John Wiley & Sons, Ltd;2012:181.
- Villani NA, Bullock G, Michaels JR, et al. A mixed breed dog with neuronal ceroid lipofuscinosis is homozygous for a CLN5 nonsense mutation previously identified in border collies and Australian cattle dogs. Mol Genet Metab. 2019; 127(1):107-115.
- Wakley SU, Suzuki K, Suzuki, K. Chapter 6: Lysosomal Storage Diseases. In: Love S, Budka H, Ironside JW, eds. Greenfield’s Neuropathology. CRC Press;2015:495-507.
- Zierath S, Hughes AM, Fretwell N, et al. Frequency of five disease-causing genetic mutations in a large mixed-breed dog population (2011-2012). PLoS One. 2017;12(11): e0188543.