Signalment:
Gross Description:
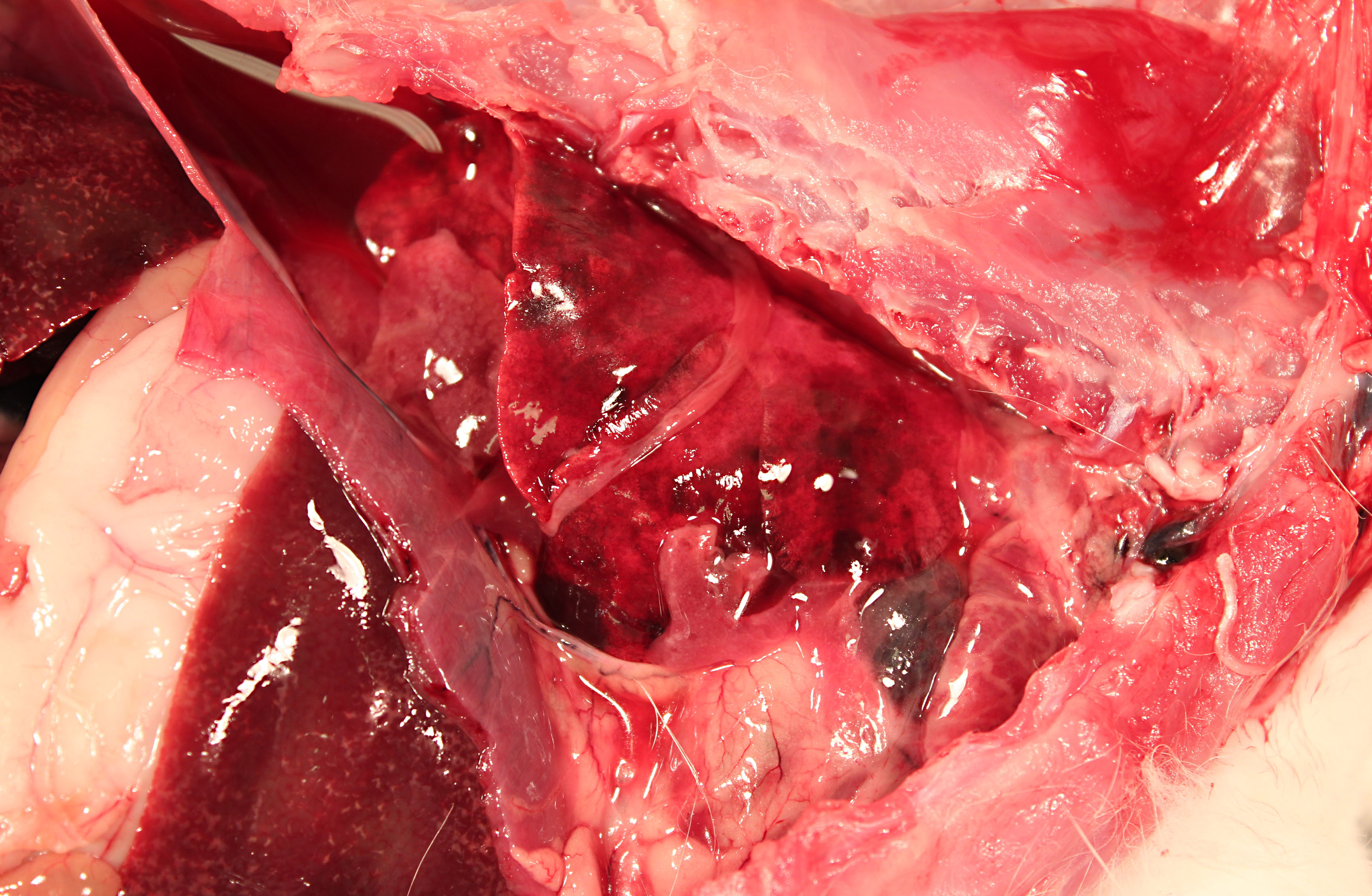
The pericardium of the heart is diffusely thick (up to 3mm wide), discolored red-tan, and rough. Approximately 1mL of yellow-red, opaque pericardial fluid is present. The epicardial surface of the heart is rough and covered with multifocal strands of gelatinous to stringy, tan material (fibrin). The endocardial surface of the heart and heart valves are grossly within normal limits.
The pleural cavity contains approximately 10mL of cloudy red-orange fluid. Dozens of 1-3mm thick strands and large coalescing mats of fibrin bilaterally cover the cranial lung lobes. Bilaterally, the cranioventral 15% of the lungs is dark red and consolidated. The remainder of the lung is mottled dark red.
Multifocally throughout the liver are hundreds of coalescing 1-2mm pale tan foci.
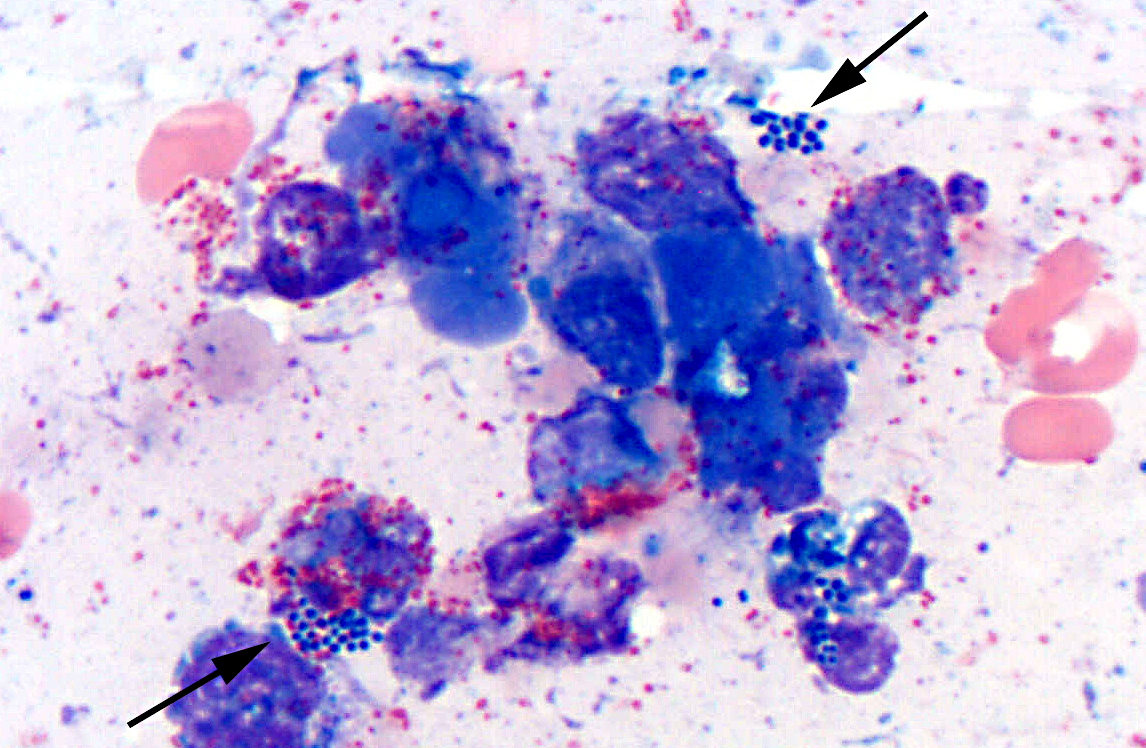
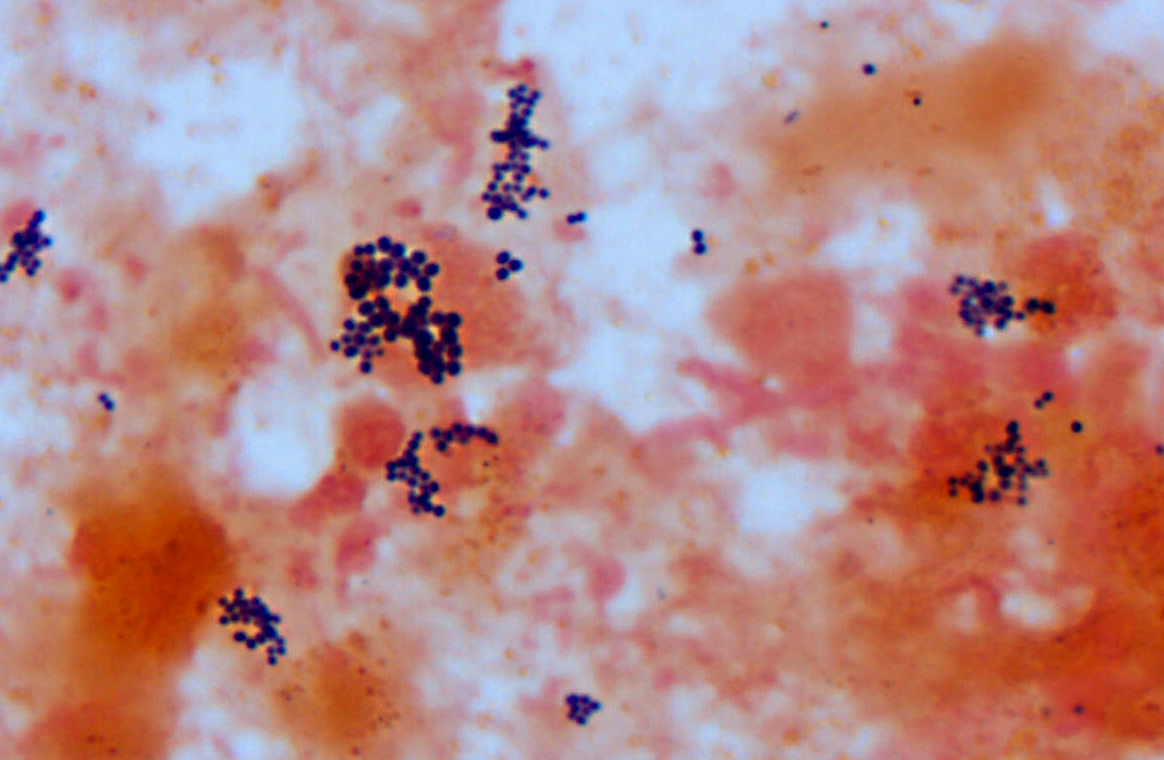
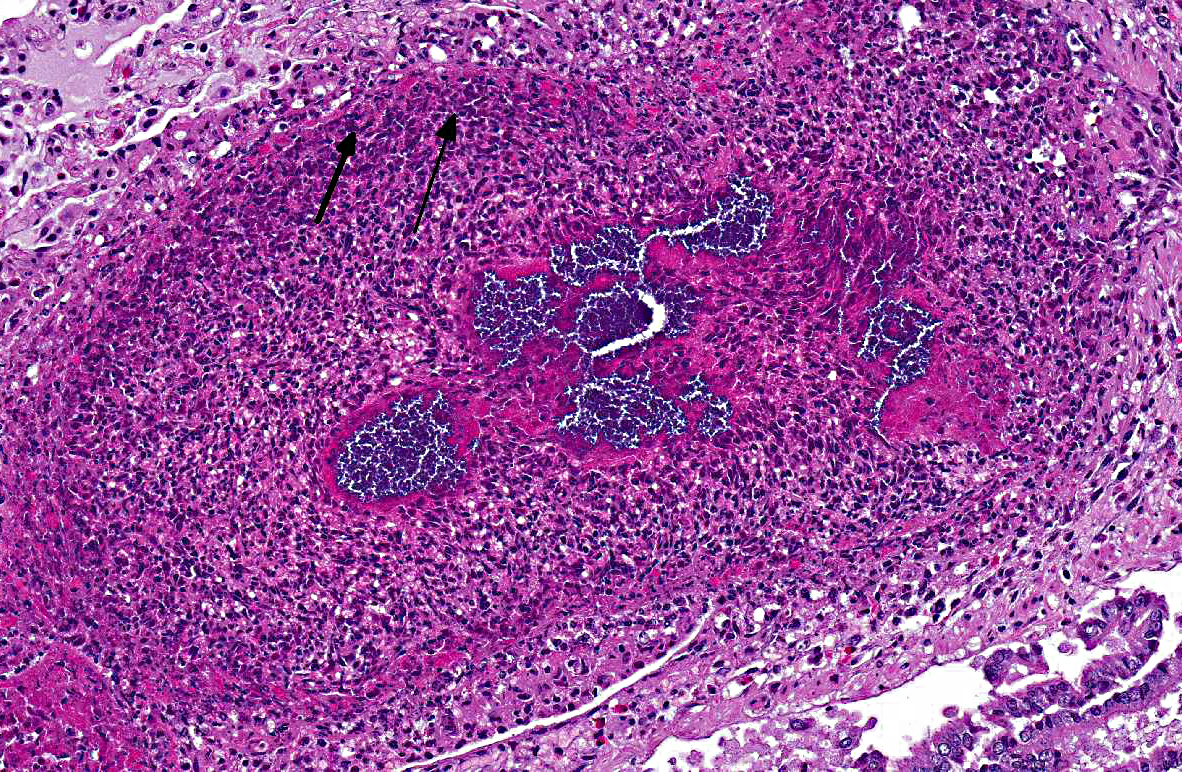
Histopathologic Description:
Morphologic Diagnosis:
Lab Results:
Condition:
Contributor Comment:
Vascular catheters are commonly used indwelling medical devices in both animals and humans. In a research setting, they are frequently used in laboratory animals to provide convenient vascular access for repeated injections with less restraint and handling stress on the animal. However, catheterization can be associated with many complications, the most serious being catheter-related septicemia.(1) Although catheter-related complications in humans are widely reported, there are very few reports of cases in animals.
Microbes that colonize catheter hubs and the skin surrounding the insertion site are the most common source of catheter-related infections.(2) Migration of skin organisms into the cutaneous catheter tract with subsequent colonization of the catheter tip is the most common route in humans.(2) Infection of the catheter hub, hematogenous seeding from a distant site of infection, or contaminated infusate are other sources of catheter-related infection.(1,2,3) Bacteria, including Staphylococcus epidermidis, S. aureus, Pseudomonas aeruginosa, Klebsiella pneumoniae and Enterococcus faecalis, and the Candida spp. of fungi are commonly involved in human catheter related infections.(1,2)
Studies have shown that virtually all indwelling central venous catheters in people are colonized by microorganisms embedded within a biofilm matrix.(2) The biofilm consists of both host and microbe-associated components. As the host reacts to the catheter, a thrombin sleeve rich in fibrin and fibronectin coats the catheter. Microbes may also produce an extracellular polysaccharide -�-�slime. Together, these form the biofilm which enhances adherence of organisms and also protects organisms from phagocytosis, antibiotics and antibodies.(1,2)
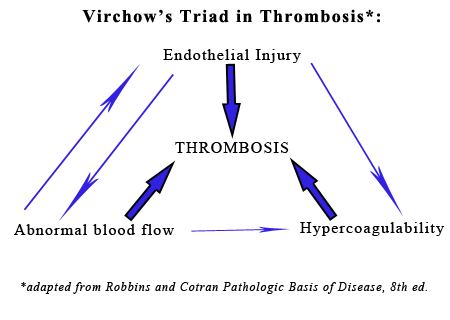
The presence of a catheter within a vessel lumen can result in endothelial injury as well as alterations in normal blood flow around the catheter, both of which predispose to thrombosis.(4) There are four possible sequelae to thrombus formation propagation, embolization, dissolution, or organization and recanalization.(4) In this rabbit, multiple emboli showered the pulmonary vessels of the lungs resulting in acute death.
JPC Diagnosis:
Conference Comment:
In contrast to these antithrombotic properties of normal endothelium, injured or activated endothelium induces thrombosis by inducing platelet binding and aggregation, activating the extrinsic coagulation cascade and enhancing the activation of clotting factors IX and Xa. Endothelial cells can by activated by trauma, infectious agents, hemodynamic forces, circulating plasma mediators, and cytokines. Endothelial injury results in exposure of the underlying extracullular matrix and subsequent adhesion of platelets to it via interactions with von Willebrand factor (vWF). vWF, which is secreted by endothelial cells, acts as a bridge between the ECM and the glycoprotein Ib receptor on platelets, allowing the formation of the primary hemostatic plug. Endothelial cells also produce tissue factor (factor III, thromboplastin), the primary activator of the extrinsic pathway of the coagulation cascade that results in secondary hemostasis. Finally, endothelial cells secrete antifibrinolytic substances such as inhibitors of plasminogen activator s (PAIs) which restrict fibrinolysis.4
The second factor in Virchows triad, abnormal blood flow (turbulence or stasis), is characterized by a disruption in the normal laminar flow of blood in which cellular elements flow centrally in the vessel, separated from the endothelium by a layer of plasma. Disturbance in this laminar flow promotes endothelial activation, enhances coagulation, brings platelets into contact with the endothelium, and prevents washout and dilution of activated clotting factors, all of which contributes to thrombosis.4
The third component of Virchows triad, hypercoagulability, can be heightened by inflammation, stress, surgery, neoplasia, pregnancy and renal disease (i.e. the loss of antithrombin III in the nephrotic syndrome). Inflammation is the most common cause of hypercoagulability, resulting in a prothrombotic state due to increased amounts of tissue factor, increased platelet reactivity, increased fibrinogen, and decreased thrombomodulin.4,5 Stress and tissue necrosis (trauma, surgery) can cause transient increases in fibrinognen. Lastly, conditions that increase platelet activation (e.g. heartworm disease, nephrotic syndrome, and neoplasia) can also contribute to increased blood hypercoagulability and thus predispose an individual to thrombus formation.5
References:
2. Leonidou L, Gogos CA. Catheter-related blood stream infections: catheter management according to pathogen. Int J Antimicrob Agents. 2010;36S:S26-S32.
3. OGrady NP, Alexander M, Burns LA, et al. 2011 Guidelines for the prevention of intravascular catheter-related infections. Healthcare Infection Control Practices Advisory Committee, Publication of the Centers for Disease Control and Prevention, 2011. http://www.cdc.gov/hicpac/BSI/BSI-guidelines-2011.html Accessed June 5, 2012.
4. Mitchell RN. Hemodynamic disorders, thromboembolic disease, and shock. In: Kumar V, Abbas A, Aster JC, eds. Robbins and Cotran Pathologic Basis of Disease 8th ed. Philadelphia, PA: Saunders Elsevier; 2010:121-127.
5. Mosier DA. Vascular disorders and thrombosis. In: Zachary JF, McGavin MD, eds. Pathologic Basis of Veterinary Disease. 5th ed. St Louis, Missouri: Elsevier Mosby; 2012:78-82.