Signalment: Adult male Sprague-Dawley rat (Rattus norvegicus).
History: This was one of a group of nine rats that had tibial fractures induced experimentally and repaired by intramedullary pinning. Ten days post-surgery, four of the nine rats developed necrotic ulcers on the ipsilateral thigh. One rat developed an ulcer on the contralateral thigh.
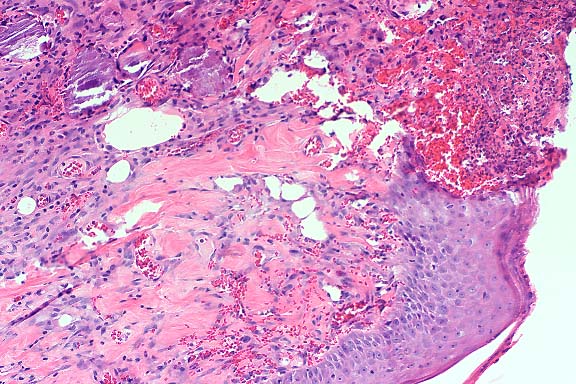
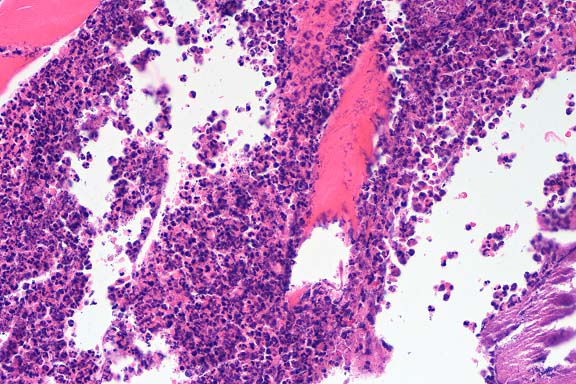
Gross Pathology: Ulcerative lesions, 2 cm in diameter, were present on the thighs of four rats. The underlying musculature was necrotic. Samples from one of the rats were submitted for histopathology and culture.
Laboratory Results: Staphylococcus aureus and Staphylococcus epidermidis were cultured from the ulcer.
Contributor's Diagnosis and Comments: Diagnosis: Severe chronic necropurulent myositis and cellulitis with granulation tissue. Etiology: Intramuscular injection of ketamine.
The specimen consists of skin and subjacent thigh muscle. Focal ulceration with severe neutrophilic inflammation is present in the epidermis. There is widespread suppurative inflammation, granulation tissue and coagulation necrosis in the muscle. Individual myofibers are swollen, hyalinized, and fragmented. Marked numbers of neutrophils surround myofibers undergoing myocytolysis and mineralization.
Conference Note: Adjacent to ulcerated areas in some sections there are moderate numbers of mixed bacteria.
Ketamine is a dissociative anesthetic agent that is frequently combined with other drugs that provide additional muscle relaxation and analgesia. The drug has been recommended for use in a wide variety of small laboratory animals and exotic species because of its wide margin of safety and ease of administration by various parenteral routes, including intramuscular (IM) injection.
Intramuscular injection of ketamine has been associated with soft tissue necrosis in the limbs of several laboratory animal species including rats, hamsters, marmosets, and guinea pigs. Administered IM, the drug causes lameness and ulceration in the injected limb that may persist for weeks. The first observed clinical signs are often self-mutilation and autoamputation of digits, especially in guinea pigs. Intraperitoneal injection (IP) of ketamine anesthetic mixtures provides an equivalent plane and onset of anesthesia with significantly less tissue damage and minimal recovery complications. Rapid dissemination and absorption of ketamine drug combinations in the peritoneal cavity may explain the lack of tissue lesions when administered IP.
With intramuscular injection, gross lesions vary depending on the dose administered and the numbers of days post-injection. In rats and hamsters at one day post-injection (DPI), subcutaneous and fascial hemorrhage, edema, and brownish discoloration of the tissue are found. At 3 DPI, pale white areas in the muscle with ulcers over the injection site are observed. Lesions become more severe with higher doses. Histologically, there is a central area of muscle necrosis characterized by sarcoplasmic swelling, hyalinization of muscle fibers, loss of striations, fragmentation, and centralized or faded nuclei. At 24 hours post-injection, a zone of inflammatory cells composed primarily of neutrophils surrounds necrotic muscle, while at three to seven days, lymphocytes, macrophages, and fibroblasts predominate. At 14 DPI, fibrous connective tissue and small foci of mineralization replace large bands of muscle.
Signalment: New Zealand white rabbit, Oryctolagus cuniculi, female, six- months-old.
History: This rabbit was used in antibody production for 3 months when it began losing weight. The rabbit had been given antigen in complete Freund's adjuvant intramuscularly (IM) initially and boosted with incomplete Freund's adjuvant. The rabbit died while being anesthetized.
Gross Pathology: The lungs contained multifocal, white, pinpoint foci on the pleural surface.
Laboratory Results: None.
Contributor's Diagnosis and Comments: Lung: Pneumonia, granulomatous, multifocal, moderate, with vacuoles (lipid).

Conference Note: Due to their size, docility, and ease of vascular access rabbits have historically been used for polyclonal antisera production. Freund's adjuvant has been used in laboratory animals many years to enhance this antibody production. Adjuvants increase the immunogenicity of antigens in two distinct ways. First, most adjuvants incorporate a substance that shields the antigen from rapid catabolism, allowing longer exposure to the host's immune system. Mineral oil is commonly used for this purpose in Freund's adjuvant. Second, many adjuvants incorporate a substance that will nonspecifically stimulate the host's antigen processing cells causing an intense inflammatory response at the injection site. The goal is to intensify the inflammatory response in order to facilitate antibody production. Complete Freund's adjuvant (CFA) incorporates killed and dried mycobacterial cells that serve as the inflammatory cell activator. Incomplete Freund's adjuvant does not include killed mycobacteria.
Complications frequently occur with use of these immunoenhancers including fever, intense local inflammation, and sterile abscess formation. Complete Freund's adjuvant has a reputation of inducing severe adverse reactions such as neuritis, self-mutilation, and skin sloughs, especially when administered serially. The lesions associated with Freund's adjuvant result from the mineral oil and, with CFA, the added mycobacteria. Such adverse reactions are usually minimized by administering the recommended dosages via the most appropriate routes and allowing for an adequate rest period between serial injections (Consult references for specific recommendations).
Both CFA and IFA have been administered intravenously to enhance
antibody production and to study the effects of injury and repair
to the lung. Multiple focal granulomas are formed subpleurally
where adjuvant emboli are often sequestered due to vascular distribution
in the lung. Type II alveolar epithelial cells become hyperplastic,
and the alveolar septa become thickened. Similar lesions may occur
with intramuscular (IM) and subcutaneous (SC) injection of CFA
and IFA. Granulomas resulting from embolism after SC and IM inoculations
may occur in glomeruli, liver, heart, and other organs. These
emboli probably result from the entrance of adjuvant into blood
vessels from necrotic tissue near the inoculation site. Lipid
containing macrophages mobilized from these sites may also become
sequestered in the lung and cause foci of granulomatous inflammation.
These pulmonary granulomas usually resolve and are not detected.
Acute respiratory distress and death in sensitized rabbits can
occur after administration of booster doses of CFA. The clinical
finding of dyspnea and post mortem finding of massive pulmonary
edema are consistent with a severe and acute hypersensitivity
reaction. Histologically, the alveoli become expanded with abundant,
eosinophilic proteinaceous fluid, and the alveolar septa are congested.
Administering only one dose of CFA, followed by administration
of IFA or antigen alone for boosters often eliminates this complication.
Freund's adjuvant causes several undesirable effects, which preclude its use in humans or in some animal species used in research. Use of CFA in nonhuman primates causes sensitization to mycobacteria and invalidation of skin testing for tuberculosis. Humans accidentally inoculated with CFA have reported chronic pain associated with severe inflammation. There may be a risk of severe allergic reactions and autoimmune disease. Uveitis, arthritis, and increased risk of neoplasia have been reported. Freund's adjuvant should be handled as a potentially hazardous substance.
Signalment: Fourteen-month-old, SJL, male mouse (Mus musculus).
History: This animal was a control mouse (did not receive any experimental manipulations) on a long-term study and was housed under conventional conditions.
SJL mice are an inbred line derived from Swiss Webster mice and have been used to study autoimmune diseases (experimental autoimmune encephalomyelitis, experimental thyroiditis, experimental interstitial nephritis, and autoimmune myositis) as well as lymphosarcoma (formerly called reticulum cell sarcoma). Ninety percent of SJL mice spontaneously develop lymphosarcoma by 12 months of age, and nearly 100% develop myositis by eight months.
Lymphosarcoma begins in mesenteric lymph nodes and Peyer's patches and has considerable morphologic variability, even at different sites within a single animal. The neoplasia is derived from a B lymphocyte but is dependent on T-cell cytokines (IFN-g, IL-2) for continued growth. A significant finding in the pathogenesis was the discovery that neoplastic B lymphocytes expressed a portion of endogenous mammary tumor virus antigen that is a superantigen for Vb16 T-cells, resulting in their stimulation. Consequently, lesions contain mixtures of neoplastic B-cells, reactive T-cells and B-cells, and activated macrophages, including multinucleate giant cells.
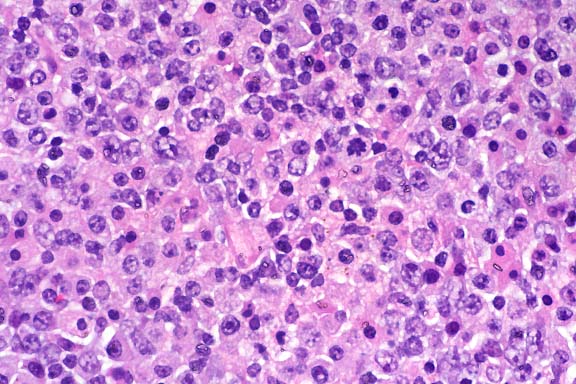
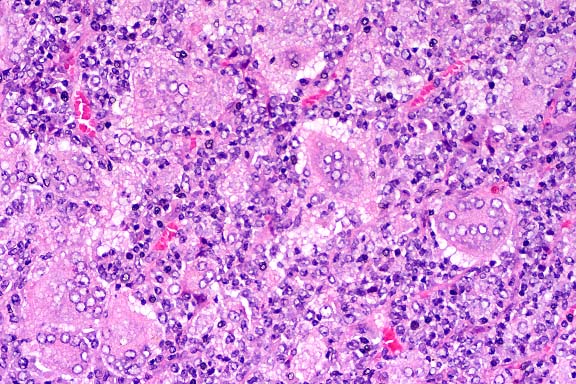
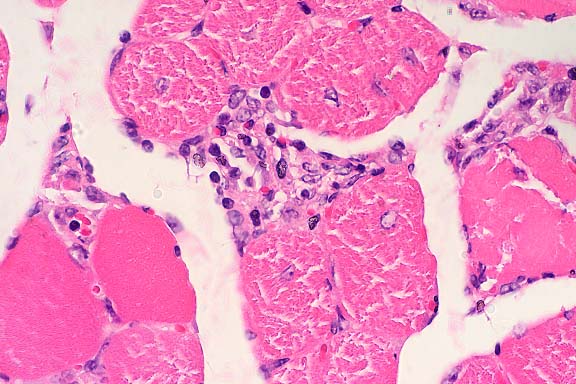
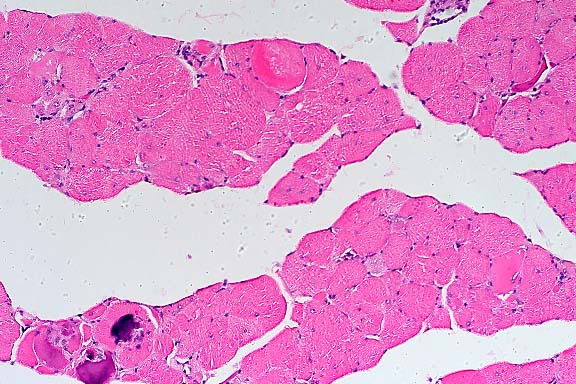
Conference Note: The SJL mouse is an inbred strain with a high incidence of spontaneous malignant lymphomas of the B-cell type often involving multiple abdominal organs. A recent study in SJL mice indicates that the histologic appearance of lymphomas from various sites may be heterogenous, even when there is clonal identity. Lymphomas arising in multiple organs within one mouse are usually derived from a single clone but may show development of subclones within a major clonal population. This finding is consistent with the concept that clonal evolution is a common event in the course of lymphoid neoplasia.
The spontaneous lymphoma in SJL mice is dependent upon host
tumor-specific immune lymphocytes for growth. The neoplastic lymphoid
cells stimulate autologous T-cells to produce lymphokines that
support the growth and proliferation of neoplastic lymphocytes.
Interleukin-5 is the most effective cytokine in stimulating growth
of tumor cells in culture. Interferon-a, IFN-b, IFN-g, and IL-2
are also induced by tumors and are important for tumor cell growth.
In addition to lymphoma, inflammatory muscle disease (myositis)
develops spontaneously in 100% of SJL mice. The various cytokines
elaborated by induced T-cells are thought to play a role in the
pathogenesis of the muscle lesions. Interferon-g, released by
stimulated T-cells during tumor growth, activates macrophages
to produce nitric oxide. Nitric oxide is synthesized by activated
macrophages under inflammatory conditions, and levels of this
molecule are elevated in SJL mice compared to control mice. Nitric
oxide functions as an inhibitor of both viral replication and
platelet aggregation and adhesion, serves as a neurotransmitter,
and is a major endogenous vasodilator. Additionally, nitric oxide
reacts rapidly with molecular oxygen and superoxide leading to
the formation of potentially toxic nitrogen oxides. While these
radicals may serve useful functions, such as the destruction of
invading organisms, they also cause collateral damage to neighboring
tissue cells, skeletal muscle for example. An inhibitor of nitric
oxide synthase, N-monomethyl-L-arginine, administered orally to
SJL mice reduces nitric oxide levels, as measured by urinary nitrate
excretion levels, and decreases the severity of the myositis.
Chronic production of elevated amounts of nitric oxide by the
SJL mouse provides a useful model for studies of cellular damage
resulting from endogenously produced nitric oxide in combination
with oxygen radicals.
Contributor: Massachusetts Institute of Technology, Division
of Comparative Medicine, Building 16-849, 77 Massachusetts Avenue,
Cambridge, MA 02139.
Signalment: Nine-week-old, female, Irish setter puppy
(Canis familiaris).
Histopath photo included with glass slide.
History: This puppy was one of six Irish setters in a litter
of 14 that developed ataxia, a wide-based stance, head bobbing,
tremors and muscular atrophy. The puppies were presented to the
Small Animal Clinic, University of Georgia at 8-weeks of age.
Within one to two weeks, they had progressed to paresis and paralysis.
Contributor's Diagnosis and Comments: Severe diffuse leukoencephalopathy with myelin degeneration, cerebrum (globoid cell leukodystrophy).
In the cerebellar and cerebral white matter, there is extensive leukoencephalopathy characterized by demyelination and massive infiltrates of large foamy phagocytic cells interpreted to be "globoid" cells. Increased numbers of vascular profiles appear rimmed with these periodic acid-Schiff (PAS) positive, Luxol fast blue positive, non-Sudanophilic, non-metachromatic cells, which are also present diffusely throughout the neuropil. GFAP immunoreactivity reveals moderate astrocytosis. Low levels of galactosylceramidase were found in all affected dogs indicating a diagnosis of globoid cell leukodystrophy.
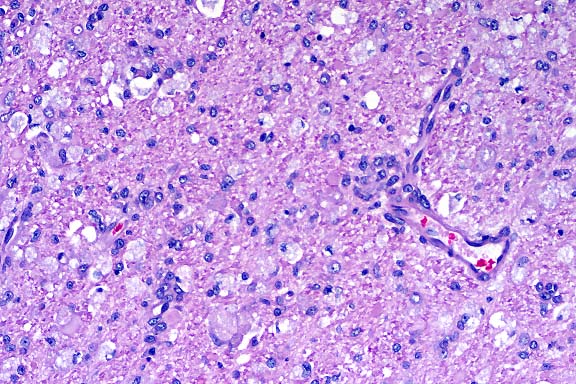

Conference Note: Lysosomes contain a variety of hydrolytic enzymes that are involved in the breakdown of complex substrates and macromolecules. With an inherited deficiency of a functional lysosomal enzyme, catabolism of its substrate remains incomplete, leading to accumulation of the partially degraded insoluble metabolite within lysosomes. Stuffed with incompletely digested macromolecules, these organelles become large and numerous enough to interfere with normal cell functions. The storage of such insoluble intermediates and metabolites occurs in cells of the mononuclear phagocyte system because they ingest and degrade erythrocytes, leukocytes, and other tissue breakdown products (e.g. myelin).
The excessive accumulation of complex substrates within the lysosomes as a result of a deficiency of degradative enzymes is responsible for a group of diseases generally referred to as lysosomal storage diseases. The numerous lysosomal storage diseases can be divided into broad categories based on the biochemical nature of the substrates and the accumulated metabolites. Within the disease categories there may be several disorders, each resulting from the deficiency of a specific enzyme.
Globoid cell leukodystrophy (also referred to as galactocerebrosidosis, galactosylceramide lipidosis, and Krabbe's disease) falls within the group of lysosomal storage diseases known as the sphingolipidoses. Inherited defects in the degradation of sphingolipids represent one of the major categories of lysosomal disease affecting humans and animals. These diseases particularly affect the nervous system and include both the classical types of neuronal storage disorders (e.g. gangliosidoses) and the disorders which primarily affect the white matter (e.g. globoid cell leukodystrophy). Sphingolipids are an important group of structural lipids in which the unifying compound, ceramide (sphingosine plus fatty acid), is esterified to one of several different molecules including sialoligosaccharides to form the gangliosides, to other saccharides to form neutral glycolipids, and to phosphocholine to form sphingomyelin.
Globoid cell leukodystrophy specifically results from a deficiency of galactocerebroside b-galactosidase, the enzyme required for the catabolism of galactocerebroside to ceramide and galactose. Galactocerebroside b-galactosidase is found almost exclusively in oligodendrogliocytes, so diseases associated with a deficiency of this enzyme manifest as leukodystrophies. The dynamic nature of normal myelin turnover results in accumulation of galactocerebroside in large mononuclear or multinuclear globoid macrophages from which the disease takes its descriptive name. Aggregates of these globoid macrophages are usually found around blood vessels within the brain or within peripheral nerves. Unlike several other storage diseases affecting the central nervous system, the deficiency of galactocerebrosidase does not cause an identifiable accumulation of galactocerebroside within either neurons or in white matter, however. Rather, an alternative catabolic pathway apparently leads to removal of a fatty acid from this molecule, leading to detectable levels of psychosin (galactosylsphingosine), a cytotoxic metabolite that is believed to cause oligodendrogliocyte injury. The resultant injury to the oligodendroglia results in myelin loss.
Grossly, the intact brain appears normal, but there is grayish discoloration and firmness of cerebral and, to some degree, cerebellar white matter. The ventricles are mildly to markedly dilated, reflecting white matter loss. White matter lesions are bilaterally symmetrical, but vary in severity within the brain and from case to case. Within the spinal cord, peripheral subpial zones are characteristically the site of lesions.
Histologically, there is gradual symmetric loss of myelin and oligodendrogliocytes in the central nervous system with a similar process in peripheral nerves. In humans, the white matter immediately beneath the cerebral cortex (subcortical U fibers) is often spared in the process, but this not a consistent feature in affected animals. Neurons and axons are also relatively spared. The unique feature of this disease is the aggregation of macrophages and multinucleated cells containing storage material around blood vessels (globoid cells).
The human variant of this disease is also known as Krabbe's disease, specifically the early infantile form. This autosomal recessive inherited disease is rapidly progressive and presents with an early onset of symptoms dominated by motor signs, including stiffness, spasticity, then weakness and decerebration. Death occurs between one to two years of age. Peripheral nerves may be involved, but no tissues outside the nervous system are affected.
In dogs, the disease has been reported in Cairn terriers and West Highland white terriers, a miniature poodle, the bluetick hound, and beagles. It has also been described in Dorset sheep, cats, and "twitcher" mice. Clinical signs in dogs begin at about three to five months of age. Often, affected dogs are ataxic and fall repeatedly on their pelvic limbs. Frequently there is also tremor of the head or a generalized tremor that is associated with activity (intention tremor). Spinal deficits progress over several weeks with loss of patellar reflexes and paraplegia, signs indicative of peripheral nervous system involvement. Muscle wasting is commonly noted. Terminally, and usually prior to one year of age, tetraplegia, terminal blindness, and dementia are observed.
Contributor: Department of Pathology, College of Veterinary Medicine, University of Georgia, Athens, GA 30602-7388.
Conference Coordinator:
Ed Stevens, DVM
Captain, United States Army
Registry of Veterinary Pathology*
Department of Veterinary Pathology
Armed Forces Institute of Pathology
(202)782-2615; DSN: 662-2615
Internet: STEVENSE@afip.osd.mil
* The American Veterinary Medical Association and the American College of Veterinary Pathologists are co-sponsors of the Registry of Veterinary Pathology. The C.L. Davis Foundation also provides substantial support for the Registry.