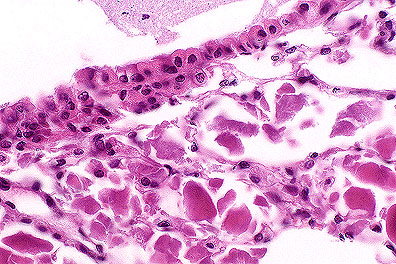
 Alveolar
proteinosis in a SCID mouse. Note the abundant eosinophilic material
in the alveoli (HE, 400X, 93K)
Alveolar
proteinosis in a SCID mouse. Note the abundant eosinophilic material
in the alveoli (HE, 400X, 93K)Signalment: 13-month-old female SCID (scid/scid) mouse.
History: This mouse was a retired breeder submitted for necropsy as part of the health monitoring program.
Gross Pathology: None.
Laboratory Results: None.
Contributor's Diagnosis and Comments: 1. Lung: Pulmonary alveolar proteinosis, multifocal, mild to moderate. 2. Lung: Pneumonia, interstitial, multifocal, mild.
In humans, pulmonary alveolar proteinosis (PAP) is an uncommon disorder in which the alveoli are filled with lipoproteinaceous material, including surfactant. It has been observed as a spontaneously occurring lesion in the lungs of CB.17 scid/scid mice and scid/scid-beige mice and resembles PAP in humans. Also, PAP has been reported in granulocyte-macrophage colony-stimulating factor (GM-CSF)-deficient C57BL/6 mice (knockout mice). Light microscopy shows varying amounts of a homogeneous to granular proteinaceous material in alveolar spaces. This material is eosinophilic by hematoxylin and eosin stain and is periodic acid-Schiff (PAS)-positive. Ultrastructurally, the material resembles surfactant. Immunohistochemical staining of lung tissue, and Western blot and ELISA of lavage fluid shows marked increases in surfactant proteins SP-A and SP-B in comparison to controls. The cause of this disease in humans and mice is unknown but appears to involve a disruption of surfactant homeostasis.
AFIP Diagnosis: Lung: Proteinaceous bodies, intraalveolar, multifocal, numerous, with mild multifocal lymphoplasmacytic interstitial pneumonia and alveolar histiocytosis, SCID mouse, rodent.
Conference Note: Pulmonary alveolar proteinosis (PAP) has been reported in many species including humans, mice, rats, hamsters, guinea pigs and goats. It occurs spontaneously and in association with many systemic diseases. Grossly, PAP presents as white to gray, round foci which frequently elevate the pleural surface of the lung. These foci often expand and coalesce with time.
There are two histologically distinct presentations of PAP. The first, or type I, occurs in humans, SCID mice, and SCID/beige mice. In type I PAP, there is accumulation of granular deposits of surfactant lipoproteins within the alveoli and little or no inflammatory response. The second, or type II PAP, is characterized by the presence of large numbers of alveolar macrophages with abundant cytoplasmic accumulations of lipoproteins. Type II PAP occurs in the rat, hamster, and guinea pig and can be induced in the rat by inhalation of silica dusts or intraperitoneal injection of chlorphentermine.
The abnormal accumulation of lipoproteins in the airways causes impaired alveolar gas exchange, leading to exercise intolerance, dyspnea, and fatigue. Pulmonary alveolar proteinosis can affect experimental results in laboratory animals, possibly confounding or invalidating studies.
The etiology of PAP has not been determined. The finding that GM-CSF deficient mice and beige mice (which have impaired natural killer cells and neutrophils due to a defect in the lysosome granule system) develop type I PAP implicates ineffective removal of surfactant by macrophages. Overproduction of surfactant by type II pneumocytes is also considered a possible pathogenesis for the development of PAP.
As mentioned, secondary PAP develops in association with many infectious agents. Tuberculosis and Pneumocystis carinii in man are known to induce PAP. PAP has recently been induced in SCID mice by infecting their intestinal tract with Candidia albicans. Exposure to certain chemicals or silica dust, myeloproliferative disorders, lymphomas, and immune deficiencies have all been linked to the development of PAP. Although the mechanism by which these agents induce PAP is not well understood, it is believed that they adversely affect macrophage function and/or increase surfactant production.
Contributor: Emory University, G70 Rollins Research Center, Atlanta, GA 30322-4510.
References:
1. Warner T, and Balish E: Animal Model: Pulmonary Alveolar Proteinosis.
A spontaneous and inducible disease in immunodeficient germ-free
mice. Am J Path 146:1017-1024, 1995.
2. Jennings VM, Dillehay DL, Webb SK, and Brown LS: Pulmonary alveolar proteinosis in SCID mice. Am J Resp Cell Mol Bio, in press, 1995.
3. Dranoff G, Crawford AD, and Sadelain M, et al: Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 264:713-716, 1994.
4. Crouch E, Persson A, and Chang D: Accumulation of surfactant protein D in human pulmonary alveolar proteinosis. Am J Path 142(1), 1993.
Signalment: 40-day-old 740 gm female crossbred/meat hybrid rabbit.
History: In a commercial rabbitry with more than 10,000 animals, an increased mortality rate was observed in rabbits shortly after weaning and moving into the fattening house. About 90% of the animals showed diarrhea for a short period before death, while the others died without clinical symptoms. Tetracycline therapy was unsuccessful.
Gross Pathology: Changes described in this animal were representative for all dissected rabbits. The animal was moderately emaciated. Stomach and small intestines were filled with small amounts of greenish-brown liquid ingesta. The content of the cecum was brownish and foamy. Additionally, an acute alveolar emphysema was observed.
Laboratory Results: Escherichia coli was isolated from the intestine. It was negative for verotoxins VT1 and VT2, cytonecrotizing factor, and P-fimbria, but was bearing the eae-gene. Serologically, bacteria were typed as 0103:H2.
Contributor's Diagnosis and Comments: Caudal jejunum, cecum: Enteropathy, acute, diffuse, moderate to severe, with numerous round to ovoid bacteria attached to luminal borders of villous epithelia; attaching and effacing Escherichia coli-infection.
Light microscopically, the complete intestine was altered with the most striking changes in caudal jejunum, ileum and cecum. Alterations appeared as flattening, disorganization, and degeneration of villous epithelia bearing numerous round to ovoid bacteria attached to the luminal brush border. Desquamation and vacuolation of epithelial cells as well as focal atrophy and fusion of villi could be observed. The lamina propria showed a slight edema and varying degrees of infiltration by inflammatory cells (lymphocytes, histiocytes, and polymorphonuclear granulocytes). Inflammatory processes were more severe in the cecum, where crypt dilation due to cellular detritus could also be observed occasionally.
Lungs, liver, spleen, kidneys, and myocardium showed an acute hyperemia. In the spleen moderate hemosiderosis, intrafollicular single cell necrosis and some neutrophils in the sinuses were found.
Ultrastructural examination of cecal villous epithelium showed multiple ovoid bacteria of a size of up to 1 x 1,5 æm, attached perpendicularly or horizontally to the luminal surface. Microvilli were totally absent. Occasionally, single bacteria were also found within the apical cytoplasm of epithelial cells.
Additionally, ultrastructural examinations using the negative staining technique revealed rotavirus-like particles in the ingesta of some animals. Rotavirus-like particles were also demonstrated ultrastructurally in villous epithelial cells. Parasitological examination revealed few coccidian oocysts in the intestine of some necropsied rabbits.
Infections with attaching and effacing Escherichia coli strains in the rabbit are well described. Most of the examinations were performed on the REDC-1 strain (Cantey and Blake, 1977; Takeuchi et al., 1978; Cantey et al.; 1981; Peeters et al., 1984). In weanling rabbits the biotype 8+, 0103:k-H2 strain is widely distributed as a highly pathogenic strain causing a mortality rate of more than 50%. Attachment of bacteria is followed by effacement of the epithelial brush borders, destruction of absorptive cells and villous atrophy (Cantey and Blake, 1977). Peeters et al., 1984, observed first attachment at 48 hours after inoculation and marked villous atrophy and desquamation of epithelial cells after six days. Reduction in digestive and resorptive capacities results in diarrhea, poor feed conversion, weight loss and mortality. Ileal permeability is increased and cecal sodium and chloride resorption is impaired.
A hypothesis on the mechanism of the attaching and effacing effect of Escherichia coli strains is published by Wolf et al. (1988). Initial bacterial adherence near the tips of microvilli is mediated by AF/R1 (adherence factor/rabbit 1) pili, while late adherence is associated with pedestal formation which occurs at the apical cell membrane after effacement of the microvilli. This implies that the AF/R1 pilus is a non-essential virulence factor. Pedestals are extrusions of the cytoplasm containing densely clustered cytoskeletal proteins, such as filamentous actin. They protrude from the apical membrane and intimately cup individual bacteria (Cantey et al., 1981; Donnenberg and Kaper, 1992; McDaniel et al., 1995).
Attaching and effacing capacities of Escherichia coli strains are correlated with the presence of a 35-kbp gene locus (LEE = locus of enterocyte effacement; the locus includes the so-called eae-genes) containing several regions encoding factors that directly participate in lesion development (McDaniel et al., 1995). One of the first factors is intimin, a 94 kDa outer membrane protein, which was formerly recognized as a product of the so-called eaeA-gene (Donnenberg and Kaper, 1992). Secretion of these factors is also controlled by the LEE locus (McDaniel et al., 1995).
Rotavirus infections are well known in commercial rabbitries. DiGiacomo and Thouless (1986) found rotavirus antibodies in about 70% of 1-2 months-old preweanlings and could isolate rotavirus from about 25% of rabbits with diarrhea and 10% of healthy rabbits in the rabbitry. Morphological changes in affected animals are only moderate. The small intestine shows fused and moderately shortened or blunted villi with slightly flattened enterocytes (Schoeb et al., 1986).
AFIP Diagnosis: Cecum and distal jejunum (per contributor): Enteritis, subacute, diffuse, mild, with mucosal atrophy and hyperplasia, epithelial degeneration and necrosis, and myriad mucosal-adherent bacilli, rabbit, lagomorph.
Conference Note: Escherichia coli produces numerous virulence factors that are responsible for enteritis in animals and man. E. coli can be placed into three groups based on the presence or absence of toxins, the method of bacterial attachment to intestinal epithelium, and the clinical lesions they induce. The three groups are the enterotoxigenic E. coli, the enteropathogenic E. coli (E. coli in this group are further categorized as attaching-effacing E. coli, verotoxigenic E. coli, or enterohemorrhagic E. coli), and the enteroinvasive E. coli.
Enterotoxigenic E. coli (ETEC) are one of the major causes diarrhea in neonatal animals and humans. To cause disease, E. coli must first colonize the gut. This is accomplished by attaching to specific mucosal surface glycoproteins by pili (also known as colonization factor antigens). After ETEC have attached and colonized the gut they produce two types of plasmid encoded toxins, heat-labile toxin and heat-stable toxin. Heat-labile toxins cause chloride loss from enterocytes with a concomitant osmotic loss of sodium and water. Heat-stable toxins likewise promote chloride loss and also induce loss of sodium bicarbonate.
Enteropathogenic E. coli (EPEC) can adhere to the mucosa without the use of pili. This group of E. coli can be further classified based on their ability to produce toxins. Not all attaching and effacing E. coli produce toxins. The attaching and effacing E. coli attach to enterocytes in a nonintimate fashion by an adhesive factor and then in an intimate fashion by the formation of a pedestal of cytoplasm that protrudes from the enterocyte. The enterocyte microvilli degenerate and are lost into the gut lumen. It is not clear how attaching and effacing E. coli cause the loss of the microvilli or induce the pedestal formation by the enterocytes; however, the process results in the development of diarrhea. A second subset of EPEC produce the cytotoxins verotoxin and shigatoxin; these are the verotoxigenic attaching and effacing E. coli. The last subset of EPEC, the enterohemorrhagic E. coli (EHEC), produce cytotoxins which induce an erosive fibrinohemorrhagic enteritis. The E. coli serotype O157 that is responsible for the hemolytic-uremic syndrome and hemorrhagic colitis in humans is a member of the EHEC group.
Enteroinvasive E. coli are not common pathogens of animals. The ability of E. coli to invade enterocytes is associated with a plasmid coding for outer membrane proteins. Replication of the bacteria within enterocytes causes ulceration and acute inflammation of the mucosa, resulting in lesions similar to those of Shigella or Salmonella.
Contributor: Institut fur Veterinar-Pathologie der Universitat Leipzig Margarete-Blank-Str. 4 D-04103 Leipzig GERMANY
References:
1. Cantey, J.R.; Blake, R.K. (1977): Diarrhea due to Escherichia
coli in the rabbit: a novel mechanism. Journal of Infectious Diseases,
135:454-462.
2. Cantey, J.R.; Lushbaugh, W.B.; Inman, L.R. (1981): Attachment of bacteria to intestinal epithelial cells in diarrhea caused by Escherichia coli strain REC-1 in the rabbit: stages and role of capsule. Journal of Infectious Diseases, 143:219-230.
3. DiGiacomo, R.F., Thouless, M.E. (1986): Epidemiology of naturally occurring rotavirus infection in rabbits. Lab. Animal Aci, 36:153-156.
4. Donnenberg, M.S.; Kaper, J.B. (1992): Enteropathogenic Escherichia coli. Minireview. Infection and Immunity, 60:3953-3961.
5. McDaniel, T.K.; Jarvis, K.G.; Donnenberg, M.S.; Kaper, J.B. (1995): A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proceedings of the National Academy of Sciences of the United States of America, 92:1664-1668.
6. Peeters, J.E.; Charlier, G.J; Halen, P.H. (1984): Pathogenicity of attaching effacing enteropathogenic Escherichia coli isolated from diarrheic suckling and weaning rabbits for newborn rabbits. Infection and Immunity, 46:690-696.
7. Schoeb, T.R.; Casebolt, D.B.; Walker, V.E.; Potgieter, L.N.D.; Thouless, M.E.; DiGiacomo, R.F. (1986): Rotavirus-associated diarrhea in a commercial rabbitry. Lab. Animal Sci., 36:149-152.
8. Takeuchi, A.; Inman, L.R.; O'Hanley, P.D.; Cantey, J.R.; Lushbaugh, W.B. (1978): Scanning and transmission electron microscopic study of Escherichia coli strain 015 (RDEC-1) enteric infection in rabbits. Infection and Immunity, 19:686-694. 9. Wolf, M.K., Andrews, G.P., Fritz, D.L.; Sjogren, Jr., R.W.: Boedeker, E.C. (1988): Characterization of the plasmid from Escherichia coli RDEC-1 that mediates expression of adhesin Af/R1 and evidence that Af/R1 pili promote but are not essential foe enteropathic disease. Infection and Immunity, 56:1846-1857.
10. Barker IK, Van Dreumel AA, and Palmer N: The alimentary system in Pathology of Domestic animals. Jubb KVF, Kennedy PC, and Palmer N eds. Academic Press Inc., 4th edition, pg. 200-212, 1993.
International Veterinary Pathology Slide Bank: Laser disc frame #2153-5, 3281, 3930, 8810-11, 11133, 12758, 18724, 20178, 20595-97, 22176-78, 22245, and 23369-70.
Signalment: 16-day-old 129 X C57BL mouse.
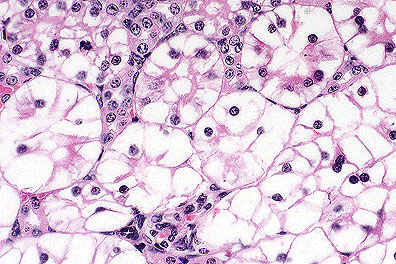 Marked tubular epithelial swelling
due to glycogen accumulation in a knockout mouse (HE, 400X, 85K)
Marked tubular epithelial swelling
due to glycogen accumulation in a knockout mouse (HE, 400X, 85K)
History: Mouse was runt in the litter.
Gross Pathology: Liver enlarged. Kidneys pale and enlarged.
Laboratory Results: None submitted.
Contributor's Diagnosis and Comments: Glycogen storage disease, liver and kidney. Glucose-6-phosphatase knockout mouse.
This mouse was developed by Dr. Hungwen Chen and Janice Chou, of National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, Maryland.
These mice also have pancreatic and bone lesions.
The purpose of the procedure to develop this knockout mouse was to simulate the human disease, Type I glycogenosis (von Gierke's disease).
AFIP Diagnosis: 1. Kidney, tubular epithelium: Vacuolar change (glycogenosis), diffuse, severe, 129 x C57BL strain mouse, rodent.
2. Liver: Vacuolar change (glycogenosis), diffuse, severe, with multifocal necrosis, extramedullary hematopoiesis, and biliary hyperplasia.
Conference Note: Glycogen storage diseases result from genetically inherited deficiencies in enzymes required for glycogen synthesis or catabolism. In man, 12 types of glycogen storage diseases have been identified based on the enzyme that is deficient. Although a slightly different clinical picture is present for each type of glycogen storage disease, they can be placed into three categories depending upon the organ system(s) most affected (hepatic, myopathic, or systemic).
Type I (von Gierke's disease) glycogen storage disease results from the deficiency of glucose-6-phosphatase. In humans, this deficiency results in hepatomegaly and renomegaly, both caused by intracytoplasmic accumulations of glycogen and lipid. People affected with von Gierke's disease also have hypoglycemia and hyperlipidemia due to deranged glucose metabolism. The hyperlipidemia predisposes those afflicted with type I glycogenosis to the formation of xanthomas and abnormal fat deposits. Type I glycogenosis has been induced in mice by X-radiation, and now by the development of the gene knockout mouse as in the submitted case. This knockout mouse develops a clinical syndrome very similar to that of human type I glycogenosis and may serve as an animal model for that disease.
Other glycogenoses have been described in animals. Type II glycogen storage disease (Pompe's disease) is caused by a deficiency in à-1,4-glucosidase and has been identified in the shorthorn and Brahman breeds of cattle; this is predominately a myopathic form of glycogen storage disease. Type II glycogen storage disease is also suspected in the cat, the Lapland dog, and Corriedale sheep. German Shepherd Dogs have been reported with a deficiency in amylo-1,6-glucosidase; this is analogous to the human type III glycogenosis (Cori's disease). English Springer Spaniel dogs have been identified with a deficiency in phosphofructokinase that is comparable to type VII glycogenosis in humans.
Contributor: National Cancer Institute NCI-FCRDC, Fairview 201 Frederick, Maryland 21702-1201
References:
1. Cotran RS, Kumar V, Robbins SL: Pathologic Basis of Disease.
W.B. Saunders Co., 5th edition, pg. 146-147, 1994.
2. Scarpelli, PG and Iannaccone PM: Cell injury and errors of metabolism, in Anderson's Pathology. Kissane, JM editor. CV Mosby Co., 9th edition, pg. 55-58, 1990.
Signalment: 4-month-old male C3H (C3H 1pr/1pr) mouse.
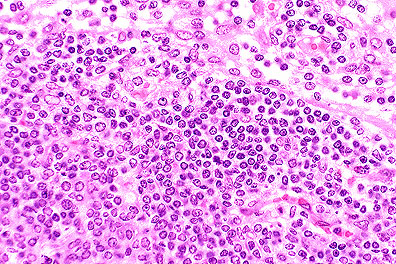 Proliferation of blastic lymphocytes
in a male C3H 1pr/1pr mouse. (HE, 400K, 127K)
Proliferation of blastic lymphocytes
in a male C3H 1pr/1pr mouse. (HE, 400K, 127K)
History: Mouse with huge lymph nodes, spleen moderately enlarged.
Gross Pathology: Large lymph nodes, generalized. Spleen - moderately enlarged.
Laboratory Results: None submitted
Contributor's Diagnosis and Comments: Lymphoproliferation disorder. (Looks like lymphoma.)
The 1pr spontaneous mutation in mice is accompanied by generalized lymph node enlargement, hypergammaglobulinemia, anti-DNA antibodies, arthritis and glomerulonephritis resembling systemic lupus erythematosus.
The nodes are greatly enlarged and composed of a monotonous population of medium-sized lymphoid cells with vesicular nuclei and sparse cytoplasm. Mitotic figures are common.
Immunohistochemically, the proliferative cells express B220 strongly (a B cell antigen) and CD3 a T cell antigen (but less so).
AFIP Diagnosis: Lymph node: Atypical lymphoid hyperplasia, diffuse, severe, C3H strain mouse, rodent.
Conference Note: The Fas antigen (Fas) is a cell-surface protein that belongs to the tumor necrosis factor/nerve growth factor receptor family. Fas is known to induce apoptosis and regulate autoreactive T-cells and cytotoxic T lymphocyte mediated cytotoxicity. FAS mRNA is abundantly expressed in the thymus, liver, heart, kidney, and ovary, but is weakly expressed in other tissues. The Fas gene is localized near the 1pr locus on mouse chromosome 19; the gene for its ligand, FasL, is located near the gld region of mouse chromosome 1.
Mice homozygous for 1pr or gld mutations do not initiate apoptosis of lymphocytes. This results in the accumulation of large numbers of nonmalignant T cells in the spleen and lymph nodes.
These mice also suffer from an autoimmune disease similar to systemic lupus erythematosus. As mentioned, Fas plays a significant role in the clonal deletion of autoreactive T-cells. Although the mechanism has not yet been elucidated, it is believed that the loss of this regulation is the cause of the autoimmune disease affecting the mice. It is possible that mutations affecting the Fas gene may cause autoimmune disease in humans.
Fas/FasL have also been shown to mediate T lymphocyte cytotoxicity in a mouse model for human fulminant hepatitis. This model utilizes transgenic mice carrying the human hepatitis B virus (HBV). Cytotoxic T lymphocytes, specific for the HBV, are injected into these mice; they induce apoptosis in the liver and a resultant fulminant hepatitis. Researchers believe that interaction of the viral peptide with the T-cell receptor activates the cytotoxic T lymphocytes and induces the expression of the gene for FasL. FasL then binds to Fas on hepatocytes and induces apoptosis. Modulation of the Fas-FasL pair could prove clinically useful in the treatment of fulminant hepatitis and other diseases caused by cell mediated immunity.
Contributor: National Cancer Institute NCI-FCRDC, Fairview 201 Frederick, Maryland 21702-1201
References:
1. Nagata S and Suda T: Fas and Fas ligand: 1pr and gld mutations.
Immunology Today, 16: 39-43, 1995.
2. Takajashi T, et al: Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell 76: 969, 1994.
3. Watanabe-Fukunaga R, et al: Lymphoproliferative disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 356: 314-317, 1992.
4. Schwartz SM and Bennett MR: Death by any other Name. Am J Path, 147:2 pg. 229-234, 1995.
Dana P. Scott
Captain, VC, USA
Registry of Veterinary Pathology*
Department of Veterinary Pathology
Armed Forces Institute of Pathology
(202)782-2615; DSN: 662-2615
Internet: Scott@email.afip.osd.mil
* The American Veterinary Medical Association and the American College of Veterinary Pathologists are co-sponsors of the Registry of Veterinary Pathology. The C.L. Davis Foundation also provides substantial support for the Registry.
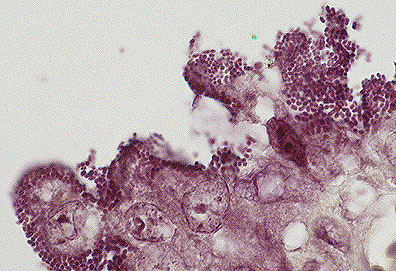 Attaching and effacing E.
coli adhered to villar epithelium in the jejunum of a rabbit.
(Brown-Hopps, 1000X, 93K)
Attaching and effacing E.
coli adhered to villar epithelium in the jejunum of a rabbit.
(Brown-Hopps, 1000X, 93K)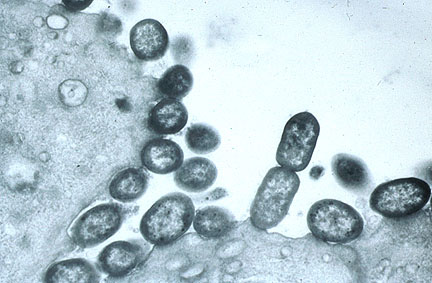 Attaching and effacing E coli
bacteria attached to the damaged enterocyte membrane. Microvili
of epithelial cells are totally effaced. (135K)
Attaching and effacing E coli
bacteria attached to the damaged enterocyte membrane. Microvili
of epithelial cells are totally effaced. (135K)