Signalment:
Over the next several months this dog developed mildly icteric mucous membranes with progression of the heart murmur to IV/VI, although otherwise remained clinically well despite persistent anemia and few crises. Three days prior to death he became icteric and lethargic and started vomiting. His clinical condition deteriorated rapidly, and aspiration pneumonia was strongly suspected. Due to a grave prognosis he was humanely euthanized.
Gross Description:
Approximately 200 ml of yellow-red serosanguineous fluid with fibrin tags was present within the abdominal cavity. The liver weighed 1070 g (6.7% of body weight), was moderately enlarged and firm, with a dark red-brown granular capsular surface and multifocal 1- 2 mm in diameter circular firm tan foci within the parenchyma. The hepatic lymph nodes were prominent with normal architecture. The spleen was dark pink and enlarged, measuring 26 x 9 x 2.5 cm. On the capsular surface, there were multifocal 2-3 mm blue to green plaques (siderofibrotic plaques).
There was 100 ml of red serosaguineous fluid in the thoracic cavity. The sternal lymph nodes were prominent with normal architecture. There was 20 ml of red serosaguineous fluid within the pericardium, and the heart weighed 180 g (1.1% of body weight). The ventral portions of the right cranial, middle and caudal lung lobes were red brown and firm. The left cranial lung lobe contained a 4-5 cm long depressed firm tan streak on the serosal surface. The right middle lung lobe sank in formalin.
Histopathologic Description:
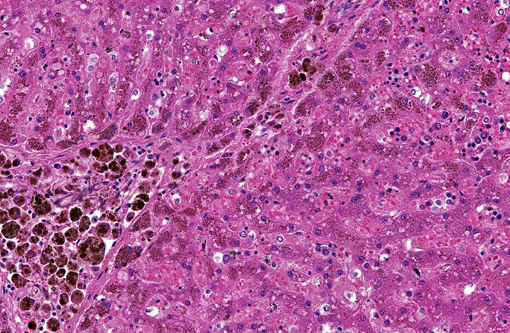
Liver (Perls iron, Figures 1A and 1B): Globular pigment within hepatocyte, macrophage and Kupffer cell cytoplasm in portal areas and extending into adjacent hepatic lobules stains positively, confirming the presence of iron.
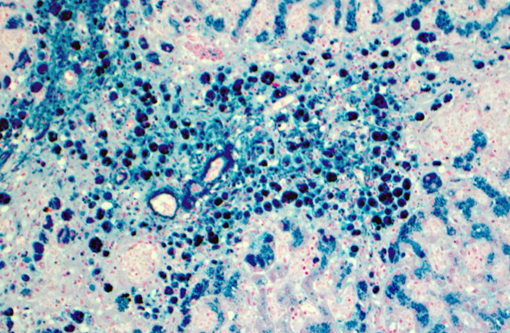
Lymph node (hepatic, Figure 2): Multifocally, medullary cords are moderately expanded by large numbers of macrophages that contain abundant dense dark brown to black globular pigment (hemosiderin), intracytoplasmic fragments of erythrocytes (erythrophagocytosis) and scattered foci of extramedullary hematopoiesis. In these regions, connective tissue and blood vessel walls often contains mineral. A mild drainage reaction is present.
Morphologic Diagnosis:
Lab Results:
- Direct antiglobulin (Coombs) test: Negative
- Erythrocytic osmotic fragility testing: Normal
- Postmortem liver iron (ICP/MS): 37,300 ppm dry weight (reference range canine: 350-1200 ppm dry weight, 100-300 ppm wet weight). Most published reference ranges are reported on a wet weight basis, dry weight results are expected to be 3.5 4 X higher than wet weight.
- DNA-based screening test for known PK mutations in Basenjis, Beagles and West Highland White terriers (8): Negative
- DNA sequencing and mutation results: Homozygous single base missense mutation of PK-LR gene (8)
Condition:
Contributor Comment:
In the dog of this report, the hemochromatosis is most likely due to the chronic severe hemolytic anemia caused by hereditary PK deficiency.(8) Pyruvate kinase deficiency is an autosomal recessive trait described most commonly in Basenjis, Beagles and West Highland White and Cairn terriers. Affected dogs present with decreased exercise tolerance, tachycardia, systolic heart murmur, pale mucous membranes and splenomegaly.(4,9) In the past the diagnosis was confirmed by measuring erythrocytic enzyme activity. This can be problematic, as erythrocytes of affected dogs lack the normal adult R-PK isoenzyme, but persistently express the M2-PK isoenzyme, which is present in many fetal and adult tissues.(9,17) Testing for known PK -LR gene mutations is simpler and now available for Basenjis, Beagles, West Highland White and Cairn terriers, and Pugs.(7,8,15) The Labrador in this report did not have any of the known mutations but was homozygous for a single base substitution in exon 7 of the PK-LR gene; this mutation renders the protein dysfunctional.(8)
Pyruvate kinase is primarily responsible for catalyzing the last step in anaerobic glycolysis. A lack of this enzyme greatly impairs ATP-generation in affected erythrocytes, resulting in decreased lifespan and an extravascular hemolytic anemia which is strongly regenerative, with a marked peripheral reticulocytosis.(9) In normal healthy Basenjis, the apparent erythrocyte half-life is approximately 10-28 days, but only 5.8 days in affected dogs.(6) It is estimated that this rapid erythrocyte turnover results in a plasma iron turnover rate that is twice normal. Prolonged and progressive iron overload frequently results in hemosiderosis and may lead to bridging portal fibrosis (cirrhosis) and thus hemochromatosis in the liver of affected dogs.(6,11,12)
Tissue is damaged by excessive uptake of iron as non-transferrin bound iron (NTBI), which, if it accumulates at sufficient concentrations, results in free radical production, iron-induced lipid peroxidation and organelle dysfunction, such as mitochondrial death.(13,14) The organs most commonly affected due to excessive cytoplasmic iron deposition include the liver and pancreas, but can also occur in lymphoid organs, as well as myocardial and skeletal muscles.(10,14,16) Fibrosis and cirrhosis develop in people at a threshold level of approximately 22,000 ppm per dry weight matter (6600 wet weight), which is 12 x normal.(10) The postmortem liver iron level in this dog was 31 x the normal upper reference range limit for dogs, resulting in the changes observed within the hepatic parenchyma.
The lesions associated with hemochromatosis occur when there is loss of equilibrium in systemic iron homeostasis, which is a fine balance between absorption and loss. Control of intestinal iron absorption is tightly regulated by multiple genes and proteins, as there is apparently no regulated mechanism for hepatic or renal excretion of iron in mammals, which occurs only through loss of body secretions, desquamation of intestinal and epidermal cells, or bleeding.(13) Absorption occurs in the duodenum and proximal jejunum across the basolateral membrane of enterocytes where it is taken up as two forms: heme iron from the digestive breakdown of hemoglobin and myoglobin, and non-heme iron released from vegetarian food sources. One of two proteins then sequester the iron to keep it non-reactive: ferritin, which stores iron in cells and is the precursor to hemosiderin, or transferrin, which is the principal iron carrying protein in plasma that distributes iron among tissues for use in biosynthesis of hemoglobin and other iron-containing proteins, and transports to hepatocytes for storage or to tissue macrophages that phagocytize senescent erythrocytes and recycle iron.(1,11,13)
Much recent work has shown that the liver plays a central role in sensing iron needs, which is modulated in part by hepcidin, an acute phase protein which is produced in the liver and secreted into the circulation.(1,3) It is thought that hepcidin responds to two independent signals, iron level and inflammatory status.(13) Hepcidin is upregulated when increased plasma iron levels are sensed, and inhibits export of iron by enterocytes and iron laden macrophages, thus participating in a negative feedback mechanism of regulation.(11,13) It functions by binding to the iron export protein ferroportin, and induces its degradation.(5) Macrophage-based cytokines IL-6, IL-1 and tumor necrosis factor-α also stimulate hepatocellular synthesis of hepcidin.(13) This mechanism is proposed to limit the availability of iron to infectious agents and tumor cells, which require iron to proliferate.(13) Decreased hepcidin expression in dogs with induced iron deficiency has recently been validated, and this work provides incentive for further investigation of gene and protein expression of this and other iron regulating molecules in the dog.(5)
JPC Diagnosis:
Conference Comment:
Osteosclerosis with the loss of marrow observed grossly in this case is consistent with other reports in dogs and does not occur in people or cats with PK deficiencies.(8) Mutations resulting in a deficiency of the enzyme phosphofructokinase, another protein in the glycolytic pathway, has also been described in dogs. It causes a persistent hemolytic anemia with occasional episodes of intravascular hemolysis due to alkalemia from hyperventilation, such as occurs during strenuous exercise. This also leads to hepatic hemosiderosis and can affect the skeletal muscle, however, osteosclerosis and liver failure has not been observed in these cases.(9)
References:
1. Andrews N. Forging field: the golden age of iron biology. Blood. 2008;112:219-231.
2. Feder JV, Gnirke A, Thomas W, et al. A novel MHC class I like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399-408.
3. Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366:348-359.
4. Fry MM, McGavin MD. Bone marrow, blood cells and system. In: McGavin MD, Zachary JF, eds. Pathologic Basis of Veterinary Disease. 4th ed. St. Louis, MO: Mosby Inc.; 2007:790.
5. Fry MM, Kirk CA, Liggett JL, Daniel GB, Baek SJ, Gouffon JS, et al. Changes in hepatic gene expression in dogs with experimentally induced nutritional iron deficiency. Vet Clin Path. 2009;38:13-19.
6. Giger U, Noble NA. Determination of erythrocyte pyruvate kinase deficiency in Basenjis with chronic hemolytic anemia. J Am Vet Med Assoc. 1991;198:1755-1761.
7. Giger U. Erythrocyte phosphofructokinase and pyruvate kinase deficiencies. In: Feldman BF, Zinkl JG, Jain NC. Schalms Veterinary Hematology. Baltimore, MD: Lippincott Williams and Wilkins; 2000:1020-1025.
8. Inal Gultekin G, Raj K, Foureman P, Lehman K, Manhart K, Abdulmalik O, et al. Erythrocyte pyruvate kinase mutations causing hemolytic anemia, osterosclerosis and secondary hemochromatosis. J Vet Intern Med. [in press, 2012]
9. Harvey JW. Pathogenesis, laboratory diagnosis, and clinical of erythrocyte enzyme deficiencies in dogs, cats and horses. Vet Clin Pathol. 2006:35;144-156.
10. House JK, Smith BP, Maas J, Lane VM, Anderson BC, Graham TW, et al. Hemochromatosis in Salers cattle. J Vet Med. 1994;8:105-11.
11. McCown JL, Specht AJ. Iron homeostasis and disorders in dogs and cats: a review. JAAHA. 2011;47(3):151-160.
12. Naigamwalla DZ, Webb JA, Giger U. Iron deficiency anemia. Can Vet J. 2012;53:25025.
13. Nairz M, Weiss G. Molecular and clinical aspects of iron homeostasis: from anemia to hemochromatosis. The Mid Europ J of Med. 2006;118:442-462.
14. Pietrangelo A. Hereditary hemochromatosis-a new look at an old disease. N Engl J Med. 2004:350;2383-2397.
15. Skelly BJ, Wallace M, Rajpurohit YR, Wang P, Giger U. Identification of a 6 base pair insertion in West Highland white terriers with erythrocyte pyruvate kinase deficiency. Am J Vet Res. 1999:60;1169-1172.
16. Sprague WS, Hackett TB, Johnson JS, Swardson-Olver CJ. Hemochromatosis secondary to repeated blood transfusions in a dog. Vet Pathol. 2003:40;334-337.
17. Stalker MJ, Hayes MA. Liver and biliary system. In: Maxie MG, ed. Jubb, Kennedy and Palmers Pathology of Domestic Animals. 5th ed. Vol. 2. Philadelphia, PA: Elsevier Saunders; 2007:308-9.
18. Tavill AS. Diagnosis and management of hemochromatosis. Hepatology. 2001;33(5):1321-1328.