Joint Pathology Center
Veterinary Pathology Services
Wednesday Slide Conference
2018-2019
Conference 12
12 December 2018
CASE II V13-12663 (JPC 4039359)
Signalment: Four year old female, Angus cow, Bos taurus,
History: The owner moved a herd of about 60 cattle from a pasture that was getting short of grass and water to an adjacent pasture with a separate water source. The environmental temperature for the time period was over 90° Fahrenheit (32° Celsius). The cattle had been on this pasture in the past. Two days after moving the cattle, two cows had died, two cows were recumbent and could not stand, and 11 head of cattle were staggering when walking. The two recumbent cows and eventually the 11 head of cattle that were staggering showed neurologic signs of blindness and ?seizures? as described by the owner. All of these cattle with neurologic signs eventually died. In total, the owner lost 15 head of cattle. The owner had lost 22 head of cattle with a similar scenario about 3 years prior to this loss of cattle. The horses on the pasture were healthy with no clinical signs of illness.
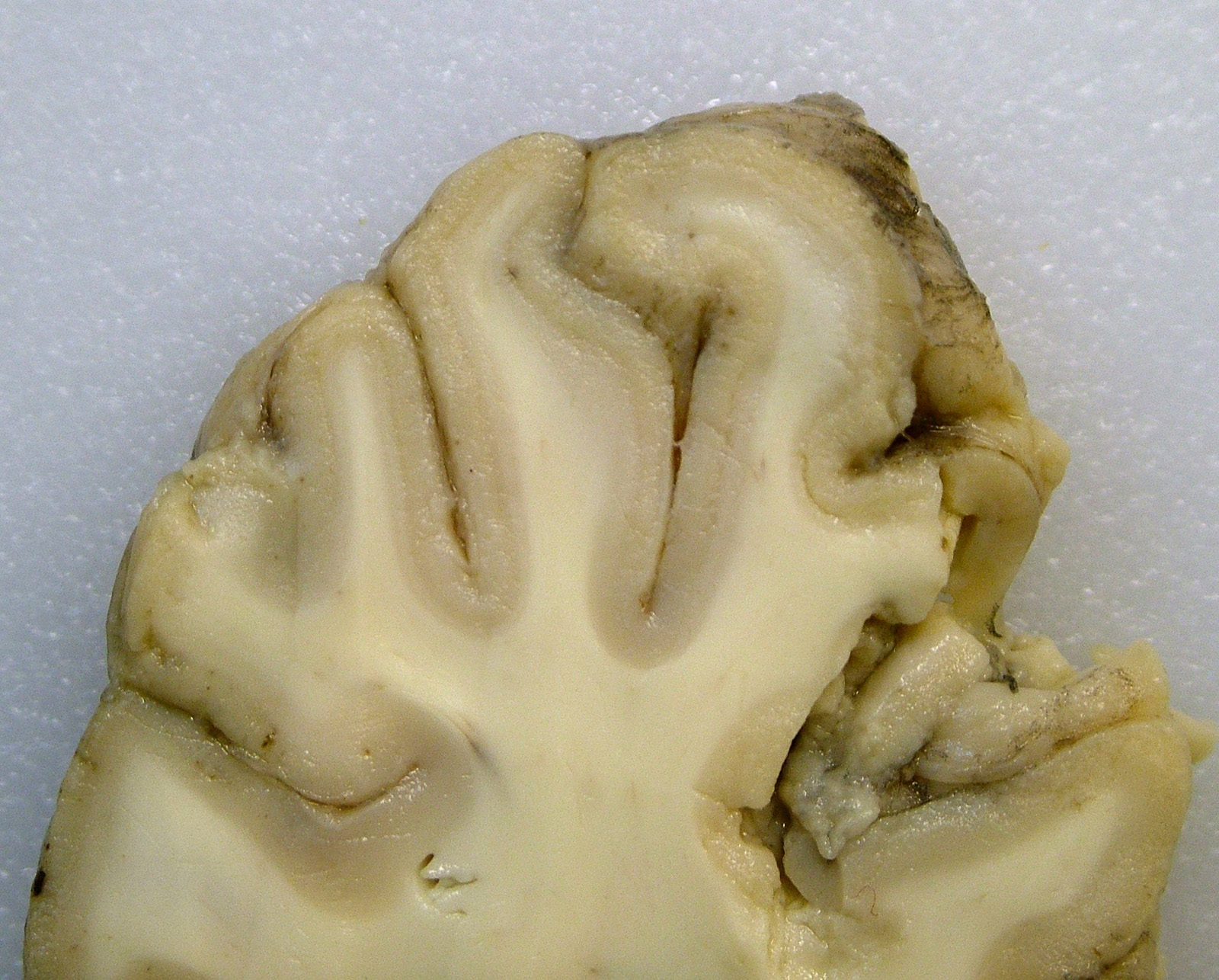
Gross Pathology: The submitting veterinarian performed the postmortem examination with no significant gross findings. He submitted fresh and fixed tissues for examination. Upon arrival at the diagnostic laboratory, the fresh tissues were severely autolyzed. In the fixed brain, there were light yellow areas of malacia in the cerebral gray matter that fluoresced when placed under an ultraviolet light with a wavelength of 365 nm.
Laboratory results: The water on the pasture was determined to be of unsuitable quality and possibly toxic for cattle with a sulfate concentration of at least 4000 mg/L (ppm), which is the upper reference limit for sulfate at the laboratory testing the water.
Microscopic Description:
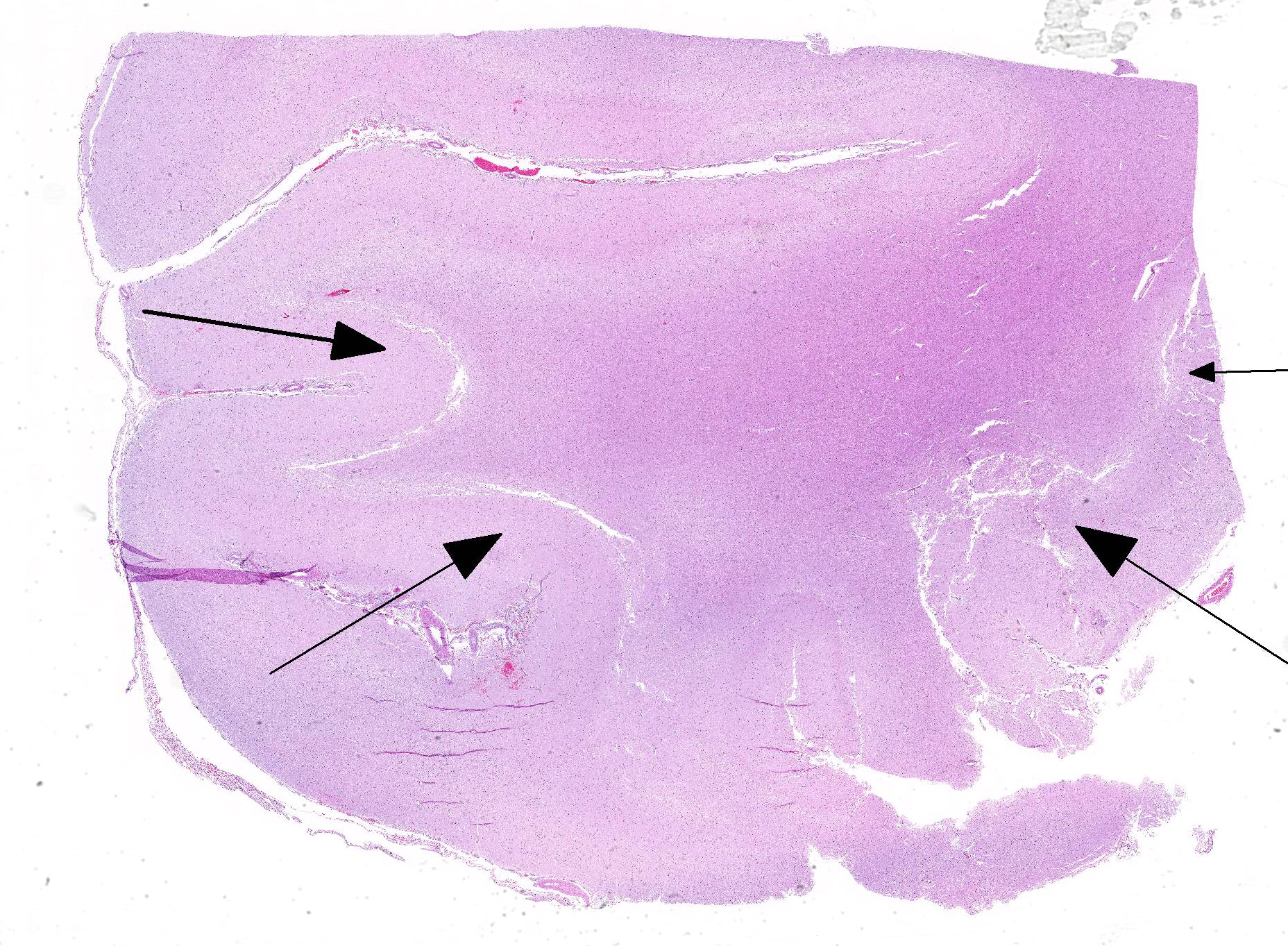
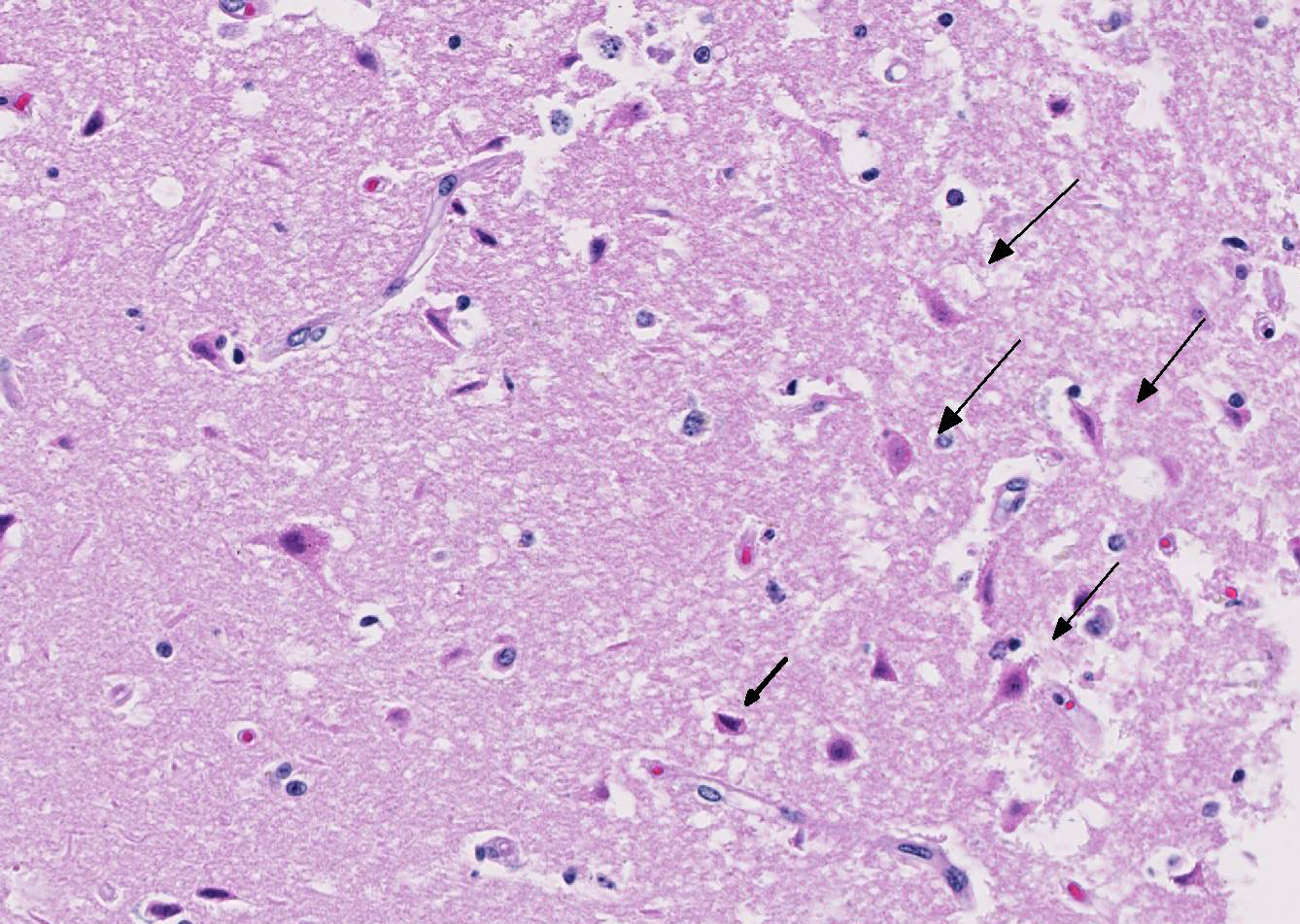
The cerebral gray matter of the brain contained multiple foci where the neuropil is pale-staining and vacuolated with prominent vasculature with swollen endothelial cells. The affected areas tend to involve the deep and middle lamina associated with the sulci, but often extend to the superficial lamina and extend up to the middle of the gyri. Within the affected foci, the neurons are often necrotic and are shrunken and hypereosinophilic with angular borders and dark pyknotic nuclei. There are occasional areas in the affected foci and associated leptomeninges where there are perivascular infiltrates of small numbers of macrophages, lymphocytes, and glial cells.
Contributor?s Morphologic Diagnoses:
Brain, cerebrum, gray matter - Polioencephalomalacia
Contributor?s Comment: Necrosis of the neurons in the lamina of the cerebral gray matter in cattle can be caused by sulfur toxicosis, thiamine deficiency, water deprivation/sodium ion toxicosis, lead intoxication, and ischemia/anoxia.1,4 The polioencephalomalacia in this case was most likely the result of sulfur toxicosis.
When calculating the sulfur content within ruminant diets, all sources including feedstuffs and water need to be considered and calculated into the total dietary consumption of sulfur.1,2 The dietary requirement of sulfur in beef cattle diets is not well defined. According to the National Research Council, the recommended concentration of sulfur in beef cattle diets is 1500 ppm with 4000 ppm being the maximum tolerable limit for sulfur in the diet.5 Thus, a 1000 pound (454 kg) cow consuming approximately 2% body weight of feed (9.08 kg) daily would have a maximum tolerable daily limit of 36.32 g of sulfur. At 90° F (32° C), it is estimated that a 454 kg cow would consume 20.6 gallons (78.0 liters) of water per day. In this case, the water contained at least 4000 mg/L sulfate. Thus, the cow consumed an estimated 104 g of sulfur in the water, which is almost 3 times the recommended tolerable daily limit of sulfur in the beef cattle diet.
The pathogenesis of how sulfur results in neuronal necrosis is not well defined. In the rumen, the excess sulfur is converted to hydrogen sulfide gas, which can be found in the rumen gas cap.1,2,4 There is evidence that sulfide inhibits cellular respiration, which could result in neuronal hypoxia sufficient enough to result in neuronal necrosis.2 However, some have theorized that sulfite (an intermediate in the reduction of sulfate) cleaves thiamine into pyrimidine and thiazole rendering thiamine inactive thereby resulting in thiamine deficiency.4
Contributing Institution:
NMDA Veterinary Diagnostic Services, http://www.nmda.nmsu.edu/vds/
JPC Diagnosis: Cerebrum: Necrosis and, cortical, laminar, multifocal to coalescing, with cavitation, spongiosis and gliosis.
JPC Comment: The contributor provides a concise yet thorough review of sulfur-induced polioencephalomalacia (PEM) in cattle. Although the first reports of PEM were published in the early 1900s, it was not until the 1970s that thiamine, or more properly its deficiency, was identified as a cause. Sulfur was first linked to PEM in the 1980s, when it was linked to the gypsum (calcium sulfate) added to cattle reactions to control feed intake would cause cerebral lesions, and its removal, decrease the incidence of PEM.5 While an interaction with thiamine was initially suspected, it is now known that PEM may be caused by the direct action of excess dietary sulfur independent of thiamine status.5
Thiamine, or vitamin B1 is a dietary requirement in carnivores (unlike in ruminants, where it is produced via rumen microbes). Deficiency in carnivores may be the result of starvation or eating a diet high in finish that contains thiamine-splitting enzymes. High levels of sulfur in the diet may also destroys thiamine after it is metabolized into sulfate. As opposed to ruminants in which necrosis is generally seen within the cerebrum, the necrosis seen in carnivores on thiamine-deficient diets is seen in the primarily seen in basal areas including nuclei of the caudal colliculi, medial vestibular nuclei and the periventricular gray matter. The term ?Chastek paralysis? applied to this constellation of lesions was coined in the 1930?s with the first outbreak of the condition in silver foxes on the Chastek fur farm in Minnesota. The outbreak, which killed a third of the farm?s population of silver foxes, was linked to the feeding of fresh fish.
Thiamine encephalopathy is also well known in human medicine, where it is referred to as Wernicke?s encephalopathy. First described by Carl Wernicke in 1881, the condition is often seen in long-term alcoholics. Wernicke?s original cases initially underwent a subacute course of fatigue, unsteadiness, photophobia, vomiting, and weight loss. Initial neurologic syndromes manifested as acute gaze palsies, somnolence, and waxing and waning levels of orientation. These patient?s rapidly deteriorated into stupor and died within 7-14 days of admission, as thiamine was not discovered either as a vitamin or as the cause of this illness, during Wernicke?s lifetime.3 The lesions of Wernicke?s encephalopathy are similar to those seen in carnivores with Chastek paralysis, with bilaterally symmetrical lesions of the periventricular and periaqueductal gray matter, and the mammillary bodies.3
Another very interesting manifestation of thiamine deficiency is that of Alaskan Husky encephalopathy.6 This lethal syndrome in Huskies is associated with a mutation in the
SLC19A3.1 gene which encodes a thiamine transporter-2 with a predominantly central nervous system distribution. This brain-specific thiamine deficiency ultimately leads to mitochondrial dysfunction and increased oxidative stress in certain areas of the brain. Affected Huskies present with seizures, altered mentation, dysphagia, central blindness, hypermetria, and proprioception positioning deficits. Cavitary lesions are present in the thalamus, and smaller lesions are often present in the putamen, claustrum, and that the junction of the gray and white matter in the cortex, brainstem, and midline cerebellar vermis.6
From a terminology point of view, the moderator reminded the group that the term ?cortex? should be applied to the cerebral gray matter and does not include the underlying white matter. There are six levels to the cortex (which are best seen in man and non-human primates).
References:
- Gould DH. Polioencephalomalacia. J Anim Sci. 1998; 76: 309-314
- Gould DH, Dargatz DA, Garry FG, et. al. J Am Vet Med Assoc. 2002; 221: 673-677
- Isenberg-Grzeda E, Hsu AJ, Hatzoglou V, Nelso C, Breitbard W. Palliative treatment of thiamine-related encephalopathy (Wernicke?s encephalopathy) in cancer: a case series and review of the literature. Palliat Support Care 2015: 12(5): 1241-1249.
- Maxie MG, Youssef S. Nervous system. In: Maxie MG, ed. Pathology of Domestic Animals. 5th Philadelphia, PA: Elsevier Saunders; 2007: 281-457
- Niles GA, Morgan SE, Edwards WC, The relationship between sulfur, thiamine and polioencephalomalacia ? a review. Bovine Practice. 2002; 36: 93-99.
- Vernau K, Napoli E, Wong S, Ross-Inta C, Cameron J, Bannasch D, Boilen, Dickenson P, Giulivi C. thiamine deficiency-mediated brain mitochondrial pathology in Alaskan Huskies with mutation in SLC19A3.1. Brain Pathol 2015; 25(4):441-453.