Signalment:
Gross Description:
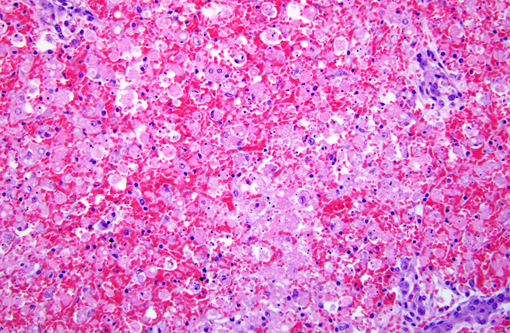
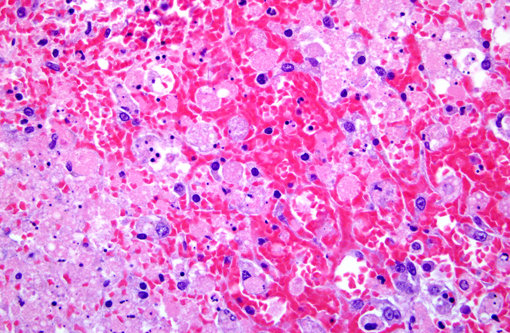
Histopathologic Description:
Morphologic Diagnosis:
Lab Results:
Severely elevated liver values (AST = 12,620; ALT = 7,540)
Coagulation times (PT/aPTT) were severely elevated (too high to measure)
Persistent and progressive hypoglycemia (initially 85 mg/dL, last measured value was 23 mg/dL)
Hypoproteinemia (Total protein = 2.5 g/dL; Albumin = 1.6 g/dL)
Acidemia (pH = 7.2)
Condition:
Contributor Comment:
Amanitins bind eukaryotic DNA-dependant RNA Polymerase II and inhibit transcription and protein synthesis, resulting in cell death. Cells with a high metabolic rate are most susceptible (intestinal crypt cells, hepatocytes, proximal convoluted tubule cells). Amanitins also result in endocrine abnormalities. (4)
Reported clinical signs:
Phase 1 Latency, the animal may appear normal for 8-12 hours following ingestion
Phase 2 Severe GI signs due to gastroenteritis (nausea, vomiting, bloody diarrhea, abdominal pain)
Phase 3 Apparent clinical improvement
Phase 4 Multi-organ failure: Fulminant liver, often with coagulopathy and encephalopathy; Acute renal failure;
Severe hypoglycemia, associated with the breakdown of liver glycogen. Coma and death typically occurs 12-84 hours following ingestion of a lethal dose. (3)
JPC Diagnosis:
Conference Comment:
Embryonic biliary precursor cells form a periportal sheet called the ductal plate, which is progressively remodeled to generate intrahepatic bile ducts. A limited number of ductal plate cells participate in duct formation; those not involved in duct development are believed to involute by apoptosis. The ductal plate gives rise to cholangiocytes lining the intrahepatic bile ducts, including its most proximal segments. It also generates periportal hepatocytes and adult hepatic progenitor cells. During early embryogenesis, there is a single-layer ductal plate surrounding the portal vein and portal mesenchyme, followed by the formation of double layered plates. In normal development, extensive resorption of the primitive bile ducts leads to the final stage, in which a network of fine bile ducts surrounds the portal vein.Â
Ductular reactions generally form a structure similar to the ductal plate, and in the pathologic response, are termed mini-ductal plates. These mini-ductal plates are composed of a small central blood vessel surrounded by a small amount of mesenchyme derived from the original Disse space, and a double layer of small cholangiocyte-like cells lining a slit-like lumen with peripheral tubular dilatation.Â
This new system classifies DRs into type 1, in which pre-existing ducts and ductules proliferate as a result of cholestasis (and is the type most commonly referred to as biliary hyperplasia by veterinary pathologists); type 2A, which is ductular metaplasia induced by inflammation; type 2B, which is ductular metaplasia induced by hypoxia; and type 3, which is the activation and proliferation of liver progenitor cells (LPC) in response to massive hepatic parenchymal loss.Â
A central tenet which underlies these categorizations is the idea of differentiation and dedifferentiation of cells, specifically LPCs. An LPC differentiates into a mature hepatocyte by sequentially evolving from keratin 19-positive (K19+) hepatoblast, to K19+ K7+ intermediate hepatobiliary cells, then K7+ intermediate hepatocytes and finally to K7 negative mature hepatocytes. In cholangiocyte metaplasia, such as in the type 2 DRs, the mature hepatocytes dedifferentiate in a reverse fashion. Both pathways rely on Jagged1/Notch signaling as well as a partnership with portal myofibroblasts, endothelial cells, and hepatic stellate cells, and the dedifferentiation of mature hepatocytes back to hepatoblasts recapitulates the embryonal ductal plate. It is hypothesized that the first layer of the mini-ductal plate is derived from this dedifferentiated K19+ hepatoblasts, and the second layer of the mini-ductal plate is from the activation of LPCs in the canal of Hering due to cholestasis or hypoxia.Â
Type 1 DR is the haphazard proliferation of pre-existing cholangiocytes due to severe cholestasis or interleukin-6 (IL-6) induction, which results in the activation of purinergic ATP receptors on cholangiocytes, which down-regulates NTPDase2 expressed on periductal portal fibroblasts that normally inhibit cholangiocyte proliferation. This physiologic response widens and elongates pre-existing ducts and ductules to accommodate the increase in toxic bile salts and reduce parenchymal damage in cholestasis or increased portal expansion by edema and inflammation. This type of structural adjustment, typically referred to as biliary hyperplasia, can be seen with the other categories of DR according to physiologic need.
Type 2A DR, which is generally periportal, is the ductular metaplasia of hepatocytes and formation of mini-ductal plates from LPCs, and results in the change of hepatocyte secretory polarity from horizontal as in canalicular to vertical as in cholangiocytes due to chronic cholestasis. This change in secretory polarity is due to the cytoplasmic keratin rearrangement from long-term exposure to bile salts, and also incites the dedifferentiation of mature hepatocytes to an intermediate K19+ K7+ hepatobiliary cell. This is accompanied by the activation of hepatic stellate cells located in the space of Disse into a myofibroblastic phenotype, which increases the production of connective tissue and the further induction of intermediate hepatobiliary cells into K 7+ cholangiocytes. Both DR type 1 and 2A are reversible.
Type 2B DR generally occurs in centrilobular areas and is result of hypoxia. This reaction is microscopically similar to type 2A and is induced by hypoxia-inducible factor 1-alpha (HIF-1α). This in turn upregulates fibrotic and vasoactive mediators such as platelet-derived growth factor-A (PDGF-A), PDGFB and plasminogen activator inhibitor-1 (PAI-1). This activate HSCs and LPCs as described in DR type 2A, resulting in progressive centrolobular fibrosis and ductular metaplastic proliferation.
Finally, type 3 DR, which occurs in this case, occurs in massive parenchymal loss. This represents the traditional notion of progenitor cell-based regeneration, and results from the bipotential proliferation of remaining LPCs, resident in the remaining canals of Hering into hepatocytes and cholangiocytes. Following massive hepatic parenchymal loss, there is a massive influx of fibrous connective tissue, and this proliferation is as described in types 2A and 2B, with a tendency toward the cholangiocytic phenotype due to a preponderance of myofibroblasts.
This ductal reaction classification scheme is beginning to gain popularity in the human pathology realm, and has value due to the inherent pathogenic implication with each category. While there is a long way to go before this type of pathogenically descriptive scheme finds favor in veterinary pathology, if at all, a system like this can be very useful. -
References:
2. Puschner Birgit, Rose, Heidi H., Filigenzi Michael S., Diagnosis of Amanita toxicosis in a dog with acute hepatic necrosis. J Vet Diagn Invest 2007; 19:312-317
3. Spoerke David G., Rumack Barry H., Handbook of mushroom poisoning: diagnosis and treatment. CRC Press, 1994
4. Vetter Janos, Review: Toxins of Amanita phalloides, Toxicon Vol. 36, No. 1, pp 13-24, 1998