Joint Pathology Center
Veterinary Pathology Services
Wednesday Slide Conference
2019-2020
Conference 18
12 February, 2020
Timothy Cooper, DVM, DACVP
Pathology Department
NIH/NIAID
Integrated Research Facility
Frederick, MD
CASE IV: 21465 (JPC 4135536).
Signalment: 18 month old male Bassett hound (Canis lupus familiaris)
History: Mild ataxia, with cardiac enlargement of uncertain origin.
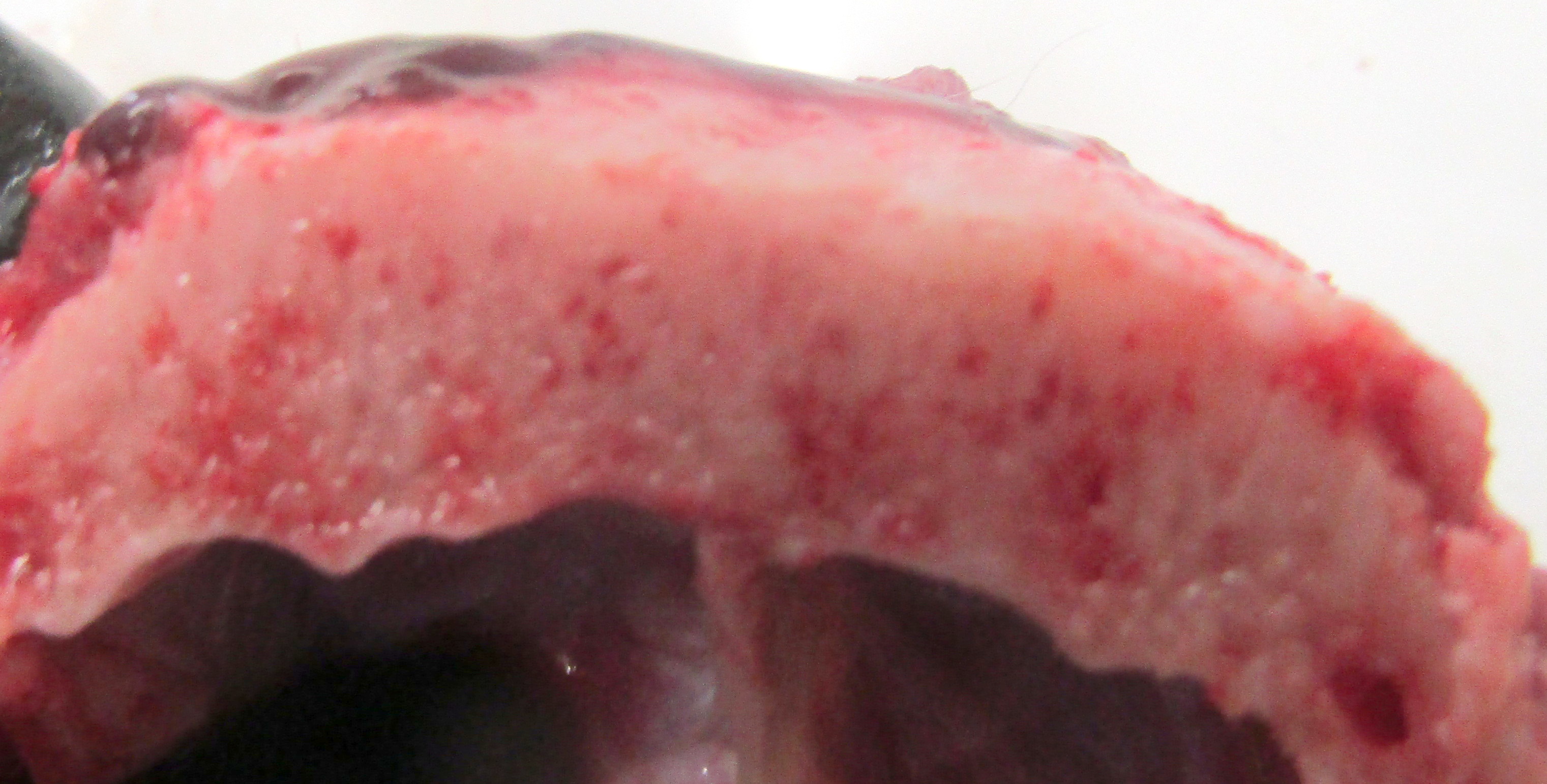
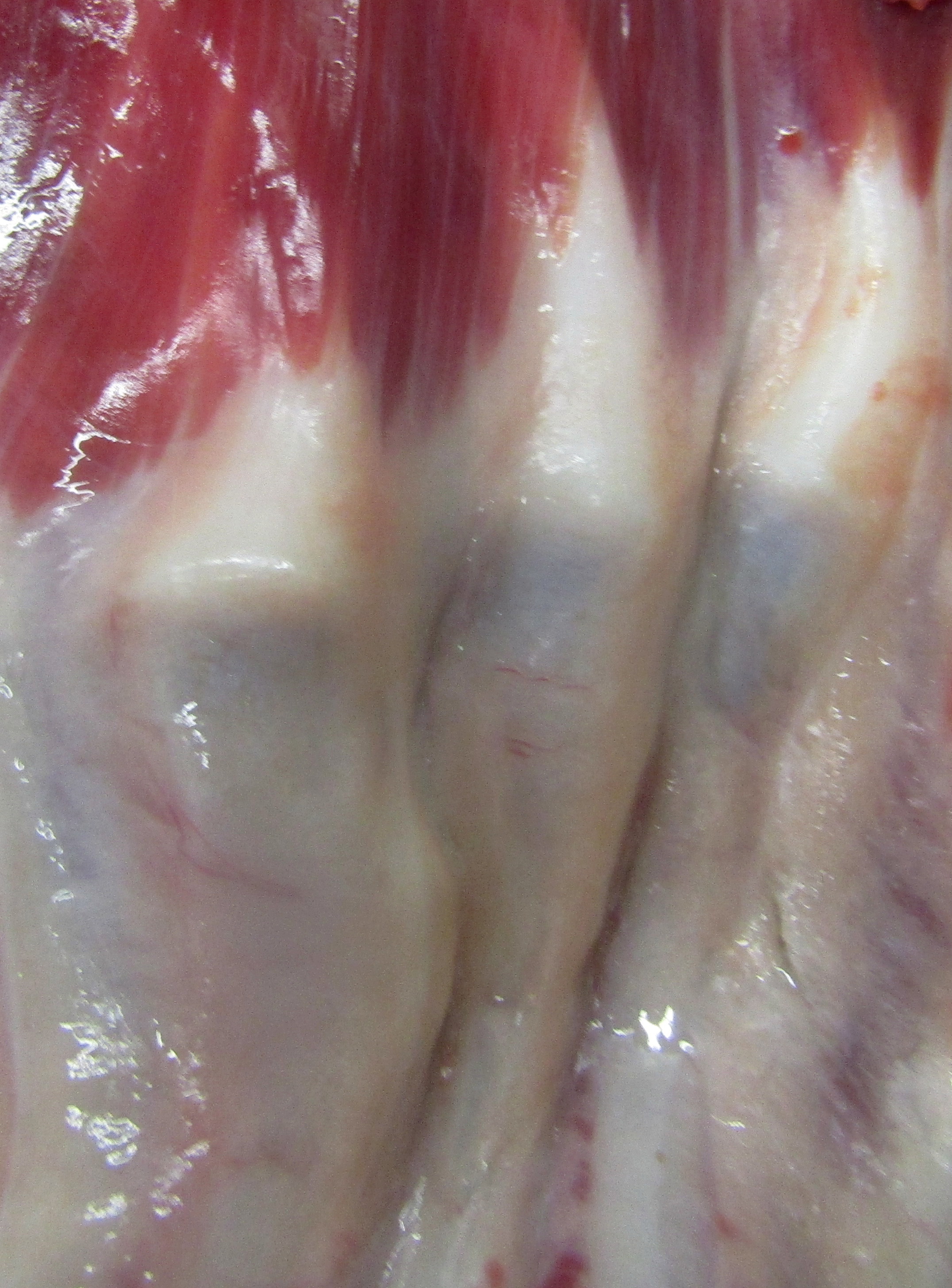
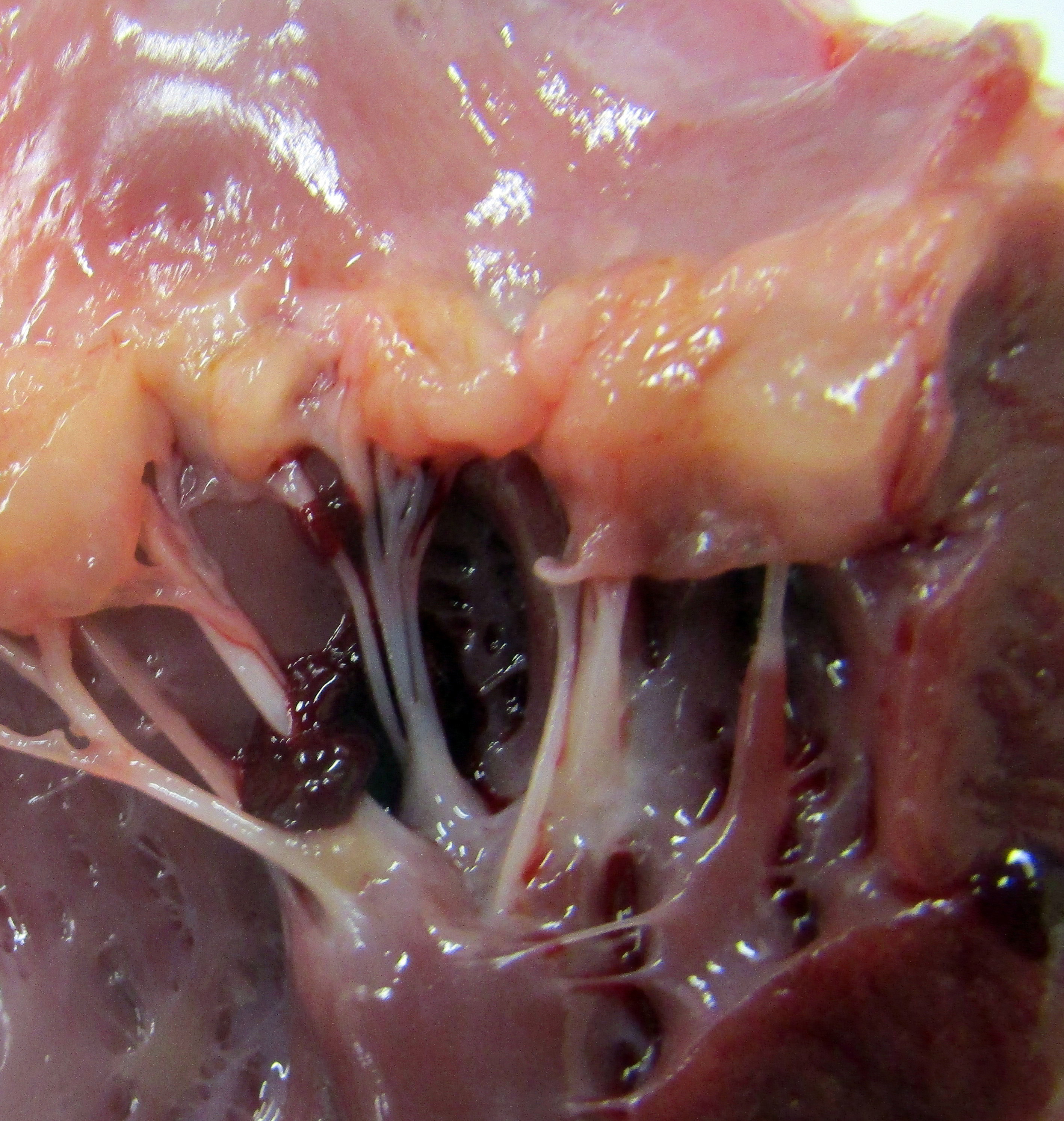
Gross Pathology: The dog has body weight of 17.0 kg. Watery joint fluid, with synovial hyperplasia, is present in several larger joints, but is particularly marked in the left coxofemoral joint. The subcutaneous fascia is thicker than expected over the muscles of the rear limbs, with reduced adipose tissue. The corneas are bilaterally cloudy. Thick dense spongy bone was present in cross-sections of the anterior calvarium The costochondral junctions of the ribs are mildly offset, with a spatulate form. The skeletal muscles are pale, especially masseters and maxillary muscles. Both interventricular heart valves are extensively and irregularly somewhat irregularly, but have, rounded, smooth surfaces, and a gelatinous texture. Associated chordae tendonae are pearly white.
Laboratory Results: A mutation was found in the α-L iduronase gene by PCR.
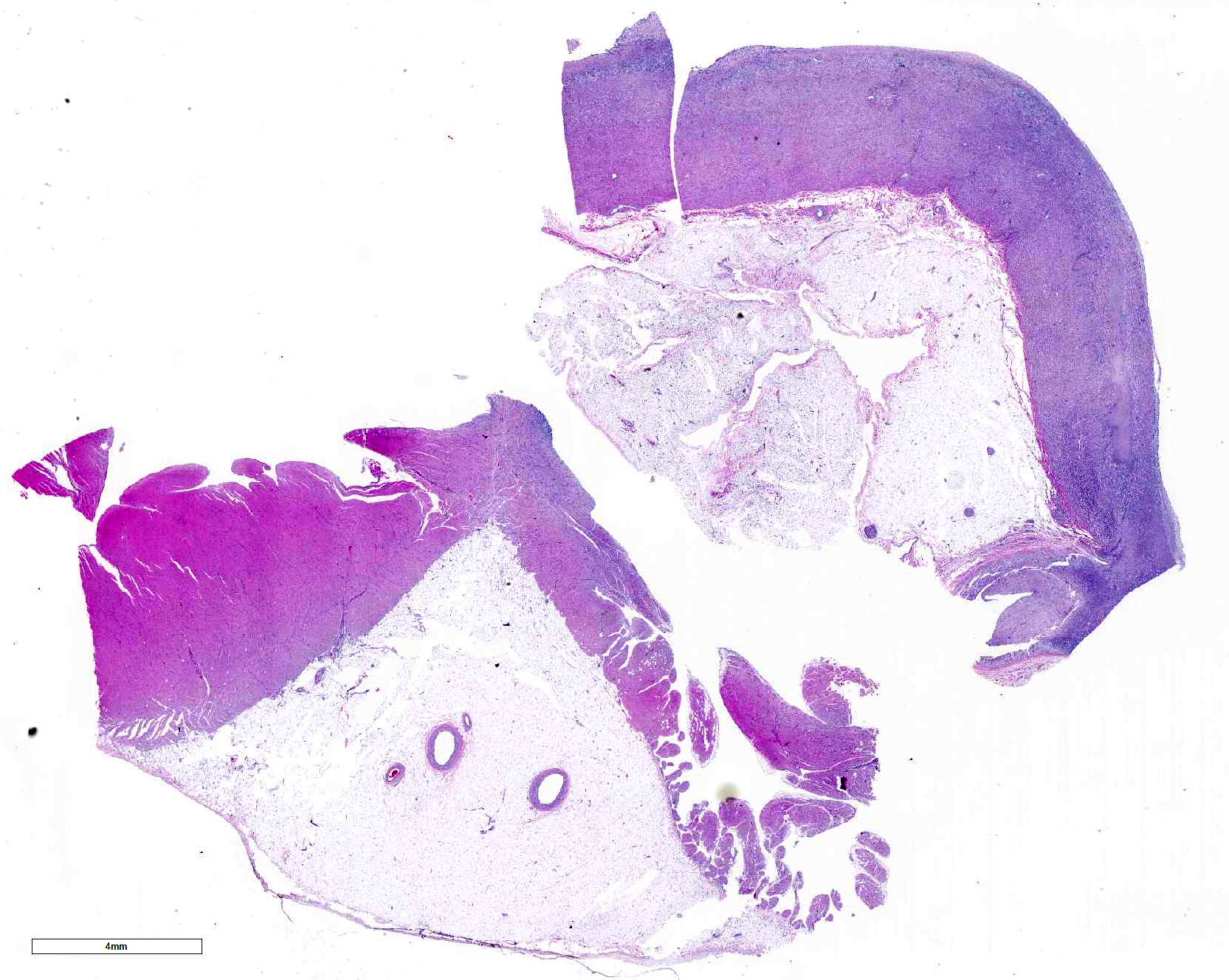
Microscopic Description: Submitted tissues consist of thoracic aorta and heart with muscular arteries in the epicardial fat.
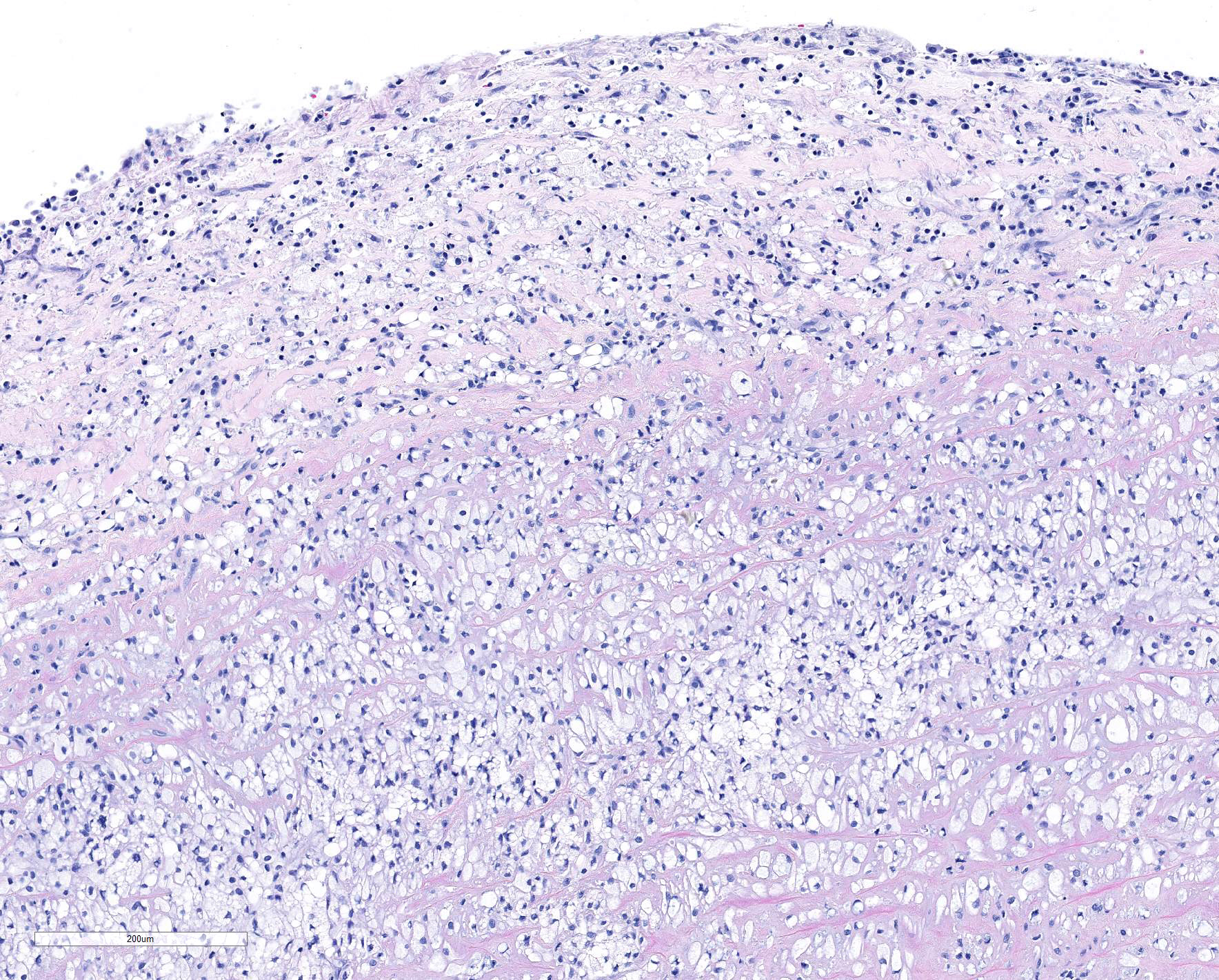
The elastic aortic wall is has elevated, plaque-like thickenings in the intima that bulge into the lumen, but remain covered by a single layer of flat endothelium. In the media, smooth muscle cells have a regimented appearance, and are separated into stacks by elastic fibers and increased interstitial ground substance with HE. Medial myocytes are diffusely contain unstained, well-defined cytoplasmic vacuoles. Despite this generally well organized microanatomy, there is multifocal disorganization of the wall in a band of vacuolated macrophages located in the intima and inner media. and multifocally, by multifocal medial vacuolation around the vasa vasorum. These areas contain a multitude of foamy macrophages that form disorganized islands in the superficial wall and contribute to the substance of the plaques. Similar vacuoles occur in the muscularis of muscular arteries in the epicardial fat with disorganization of the vascular wall. The adventitia of these arteries contains increased ground substance and vacuolated macrophages in both muscularis and adventitia. There is very mild increase in lipofuscin on either side of cardiomyocyte nuclei sectioned longitudinally (not shown).
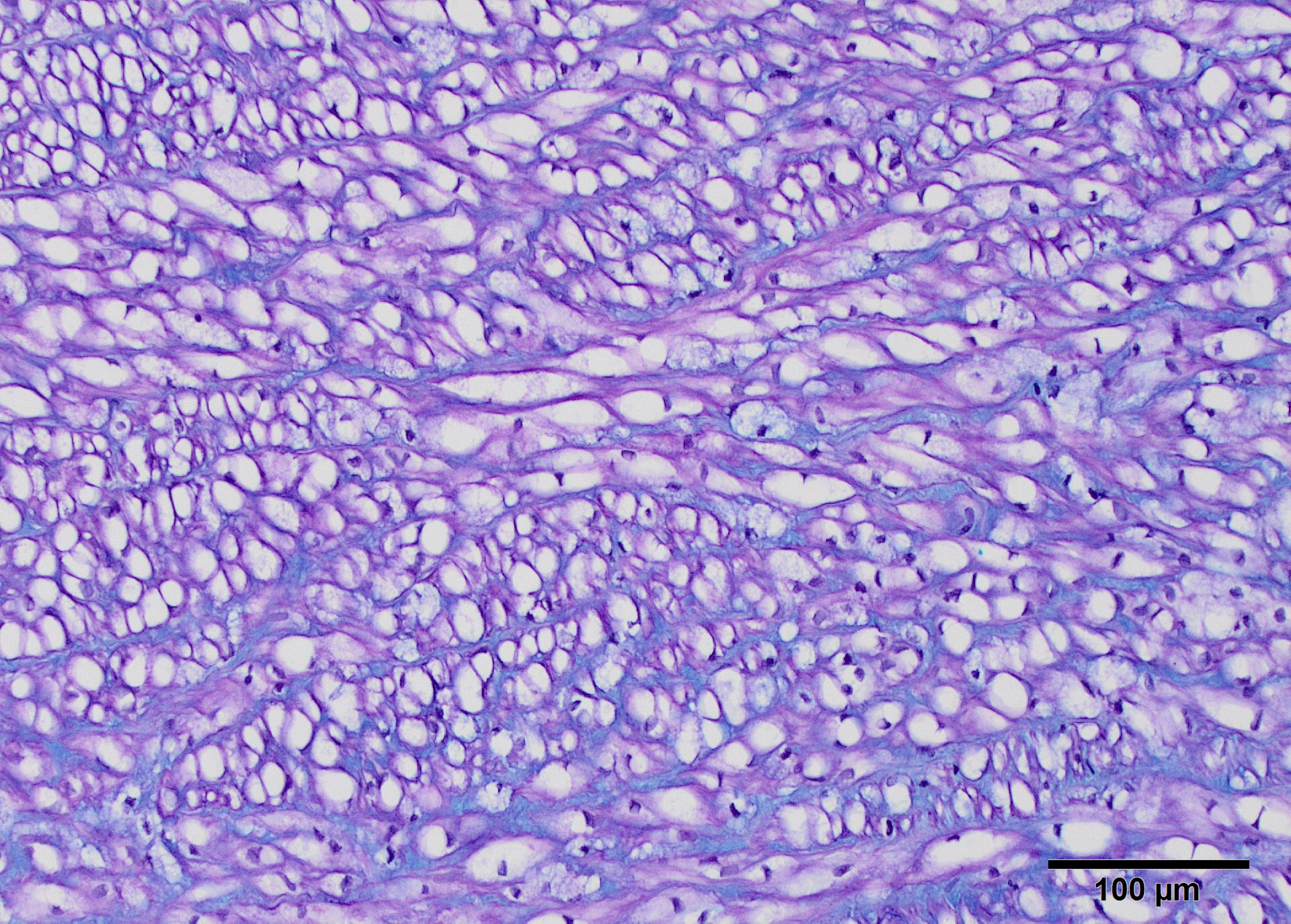
Movat?s pentachrome staining was used to differential stain to determine the stromal components of the plaques (Fig.9). This stain produces black staining of elastin, while mucopolysaccharides stain green, collagen yellow, and muscle stains red. Small elastin fibers of the tunica intima is disorganized, and the plaque is easily demarcated from the internal elastic lamina of the media. Myocytes are outlined by elastin as well. The plaque is less intensely stained green, detecting mucopolysaccharides in its ground substance. Elastin staining is less organized in the intimal plaque and around the vasa vasarum. With Alcian blue-PAS 1.0 and 2.5 blue staining of sulfomucins predominated between the medial myocytes (Fig 10), but cytoplasmic vacuoles are shaded in pink along the edges (neutral and carboxymucins mucins). Vacuoles are found within muscle cells of the epicardial arteries.
Laboratory Results (clinical pathology, microbiology, PCR, ELISA, etc.):
A mutation was found in the α-L iduronase gene by PCR.
Contributor Morphologic Diagnosis:
Arteriopathy, with aortic plaques and stromal dysplasia, consistent with mucopolysaccharidosis
Contributor Comment:
As a group, the mucopolysaccharidoses result from defective glycosaminoglycan catabolism. 9, 10 They are characterized by accumulation of glycoaminoglycans (GAG) in lysosomes. The most severe form, a deficiency of α-L iduronidase, causes Hurler?s syndrome (MPSI), and results in increased retention of both dermatan sulfate and heparin sulfate in cells and interstitium.9 Both metabolites are shed in urine of patients. Failure to hydrolyze these two substrates results in their accumulation in lysosomes, triggering complex of intracellular events leading to clinical signs that are directly linked to mechanical consequences of GAG storage.5 Common findings in humans include laryngeal and tracheal narrowing, hearing and visual deficits, gargoyle facies, skeletal deformities and heart valve disease with cardiomyopathy due to cardiac rigidity.9,10
Over 100 enzyme defects have been described in people of severe or intermediate phenotype.10 α-L iduronidase deficiency has been described in dogs 3, 11, 12 knockout mice7, 2 and cats.4 Even after replacement therapy, there is over a double increased expression of lysosomal and proteasome function in 19-month-old Plott hounds affected by MPSI.2 Downregulated were genes associated with cell adhesion, cytoskeleton and calcium regulation. Immunoregulatory genes were also elevated. This could mean that GAGs produce an inflammatory environment that exacerbates disease.2 Up-regulation of extracellular cathepsin B may also increase pathology due to elastin catabolism.6
The known mutation in Plott hounds is G to A point mutation in the donor splice site of intron, 4, 8 resulting in retained transcription of intron, but premature termination at the intron-exon junction due to the presence of a stop codon. Homozygous recessives completely lack the enzyme protein or its activity8 Mucopolysaccharidosis I has described in a Rottweiler, Afghan hound, this Bassett and Boston terrier, but information about the specific genetic mutations in these breeds is not known.
Enzyme replacement abrogates part of the clinical syndrome and increases the survival time in humans and dogs, but patients still have a considerable residual disease burden.1, 10 Enzyme replacement does not correct heart valvulopathy, cardiac stiffness of vasculopathy, and death from cardiac manifestations of disease is common, even after treatment. Bone abnormalities remain, and may require surgical intervention. Dogs given genetically corrected hematopoietic stem cells had increased GAG clearance, reduced GAGs in the brain and improved craniofacial appearance. It is noted that people with severe gene mutations like W70X, Q70X and missense A327P, G51D have no functional enzyme activity and the authors indicated that they must be referred for therapy shortly after birth to slow the progression of disease.10
Histologically, affected dogs have unstained vacuoles are present in many organs, particularly mesodermal tissues such as fascia, cartilage, blood vessels, heart valves and cerebral leptomeninges.11
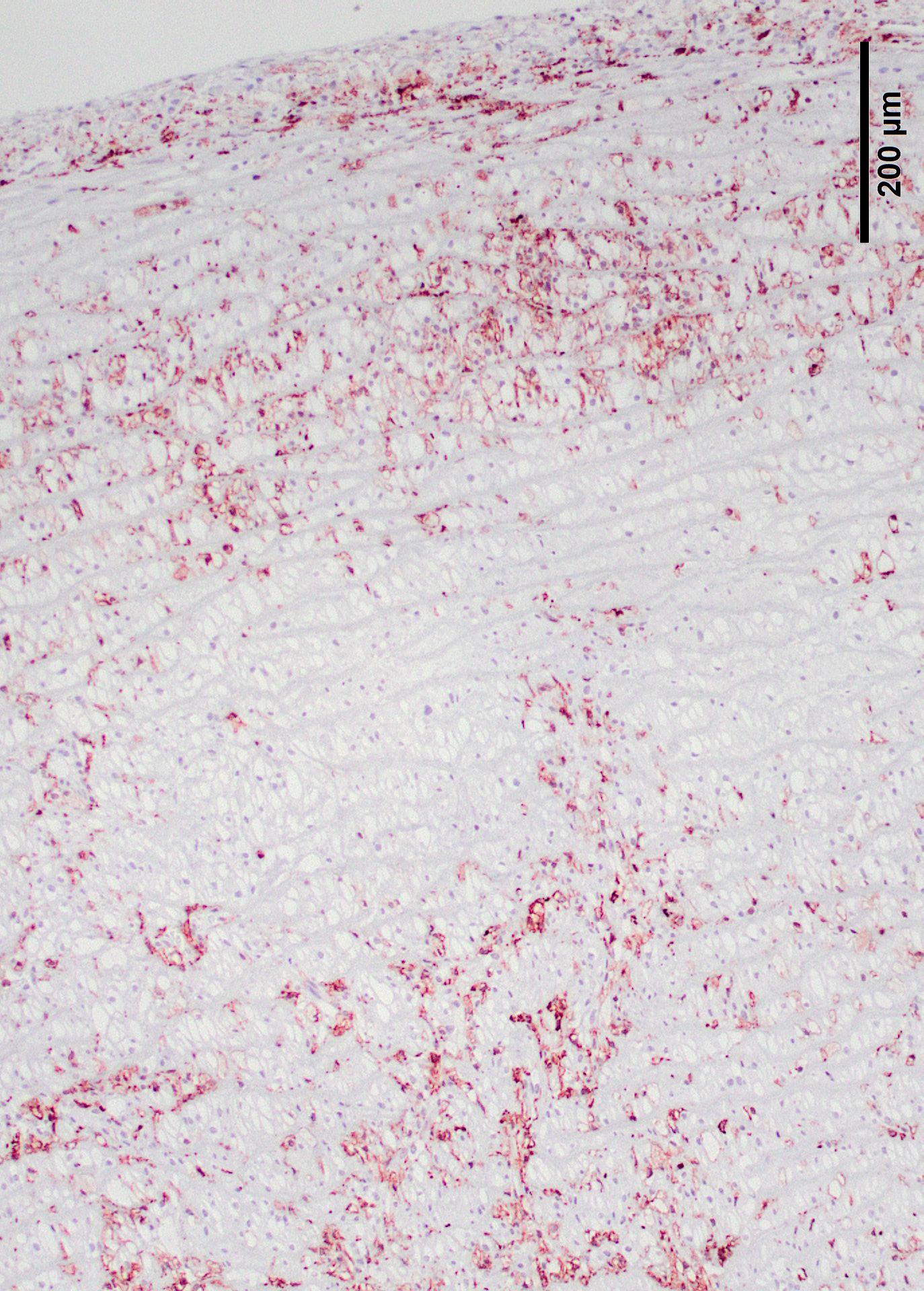
Lyons examined the branches of the terminal aorta of MPSI-affected dogs that lacked α-L-iduronase enzyme activity. 7 Vascular wall thickness is particularly increased near branch points, locations associated with turbulent flow. Reduced shear stress alters the metabolism of endothelial cells, resulting in increased permeability and leakage. Macrophages may also contribute to degradation of internal elastic lamina, as demonstrated in this case.7 These asymmetric plaques are characterized by extensive intimal thickening and disruption of the internal elastic lamina, often with significant (60-70%) narrowing of the vascular lumens. Lumenal narrowing was probably more significant in the cases in this study compared to the submitted tissue, because only the terminal branches of the aorta were used in the study. The plaques contained GAG-laden macrophages, fibroblasts and smooth muscle cells, with loss of the endothelial basement membrane and reduce claudin production.7 CD18+ macrophages were scattered or clustered below the endothelium (Figs. 6,7). Enzyme treatment makes intimal plaques more organized, with reduced macrophages and more laminar organization of fibrous tissue in treated dogs after several months. Similar pathology has been found in human patients and null mice.1 In affected humans, the disease most commonly affects the coronary arteries. In mice, dilation of the aortic root was more common and coronary artery disease was uncommon, species differences exist in disease manifestations. This progressive vascular changes in MPS-I shares some morphological similarities to atherosclerotic plaques.
Contributing Institution:
Veterinary Medical Diagnostic Laboratory, University of Missouri, 610 East Campus Loop, Columbia MO 65211, http://vmdl.missouri.edu/; http://vpbio.missouri.edu/)
JPC Diagnosis: Aorta: Smooth muscle vacuolation, diffuse, severe, with multifocal necrosis , intimal histiocytic plaque formation , medial neovascularization, and fibrosis.
JPC Comment: The contributor has down an excellent job in describing the mucopolysaccharidoses in the dog, and also in man.
The
mucopolysaccharidoses are a group of lysosomal storage diseases arising from a
genetic deficiencies of enzymes needed to catabolize the mucopolysaccharides, a
group of glycosaminoglycans constructed by attaching long-chain carbohydrates
to proteins. Most of these diseases were first described in humans in the
1970?s, and a number of animal models of spontaneous disease have been
subsequently identified (Table 1). They are most commonly found in the
interstitial ground substance and include dermatan sulfate, chondroitin sufate,
keratan sulfate and heparan sulfate. These undegraded or partially degraded
glycosaminoglycans accumulate in lysosomes in a number of cells, including
macrophages and long-lived post-mitotic cells such as fibroblasts and myocytes.
While the accumulation of partially degraded mucopolysaccharides within
lysosomes appears histologically to be a relatively benign process largely
resulting in cytomegly, the buildup of these products within lysosomes results
in tremendous enlargement and overpopulation of these organelles, resulting in
hindrance of normal cellular processes, leading to cell death. In addition,
the accumulation of autophagic substrate within lysosomes may proceed to a
level in which apoptosis may be triggered.
MPS
1, or Hurler?s disease is considered a prototypical, and one of the the most
severe forms of mucopolysaccharidoses. It results from a deficiency of α-L-iduronidase,
required to hydrolyzed the terminal α-L-iduronic acid residues from
dermatan and heparan sulfate. In humans, three distinct clinical
manifestations of the condition exist: MPS I H or Hurler?s syndrome, MPS I S
or Scheie syndrome, or the intermediate phenotypes, collectively referred to as
MPS I H/S or Hurler-Scheie syndrome.9 MPS I H is the most sevre,
with an early onset (most cases are diagnosed between 6 and 24 months of age)
and including marked cognitive delay as one of the symptoms. It is a
progressive disease which untreated, results in death before the 10 year, and
includes elarged lver and spleen, skeletal deformites, coarse facies, and joint
stiffness. Affected individuals develop significant hearing loss, and an
enlarged tongue, along with congnitive delay relegates affected individuals to
only rudimentary language skills. Communicating hydrocephalus often develops
at 2-3 years of age. MPS I S, Scheie syndrome, on the other end of the severity
spectrum is a much more limited phenotype, sharing joint stiffness (an early
and often presenting sign), aortic valve disease, corneal clouding and other
ocular abnormalities, but not the cogniftive delay, and diagnosis is often made
between 10-20 years of age.9
Treatmentof Hurler?s disease has advanced dramatically in recent years, and
include allogeneic hematopoietic stem cell transplants (HSCT) and enyme
replacement therapy with human recombinant laronidase.10 HSCT is
considered the most appropriate treatment for MPS I H, and has been shown to be
successful in halting progression of cognitive delay when jused in patints
before 2.5 years of age. Enzyme replacement therapy has been shown to be
effective in cases of MPS I S and may be initiated in more severely affected
individuals prior to more definitive HSCT; however, it is not recommended for
Hurler?s syndrome due to the limited ability of laronidase to cross the
blood-brain barrier.10
|
Disease |
Storage product(s) |
Deficient enzyme(s) |
Species |
Breed(s) |
|
MPS type I ?Hurler?s disease? |
Different glycosaminoglycans (dermatan sulfate, heparan sulfate, keratin sulfate, chondroitin sulfate) |
α-L-iduronidase |
Dog, cat |
Plott hound Domestic shorthair |
|
MPS type II ?Hunter syndrome? |
Iduronate-2-sulfatase |
Dog |
Labrador retriever (1) |
|
|
MPS type III A, B, D ?Sanfilippo syndrome? |
Heparan sulfate sulfamidase (SGSH; MPS type IIIA), a-N-acetylglucosaminidase (NAGLU; MPS type IIIB), and N-acetylglucosamine-6-sulfatase (GNS; MPS type IIID) |
Dog, cattle, goat |
IIIA ? Dachshund, New Zealand Huntaway dogs IIIB ? Schipperke dogs, cattle, emus IIID ? goats |
|
|
MPS type VI ?Maroteaux-Lamy disease? |
ARSB (arylsulfatase B) |
Dog, cat |
Miniature Pinscher, Miniature Schnauzer, Toy Poodle, Welsh Corgi, Chesapeake Bay Retriever, Great Dane Domestic shorthair, Siamese |
|
|
MPS type VII ?Sly disease? |
Β-glucuronidase |
Dog, cat |
German Shepherd Dog Domestic shorthair |
Table 1. Select mucopolysaccharidoses in animal species.
The moderator noted the presence of small vessels within the inner tunica media. These are considered abnormal (dogs should not have them in the inner half of the thoracic aorta and evidence of neovascularization and the chronicity of this particular lesion.
References:
1. Braunlin E, Mackey-Bojack S, Oanoskaltsis A, et al. Cardiac functional and histopathologic findings in humans and mice with mucopolysaccharidosis type I: implications for assessment of therapeutic interventions in Hurler syndrome. Pediatr Res. 2006;59:27-32.
2. Gonzalez EA, Martins GR, Tavares AMV, et al. Cathepsin B inhibition attenuates cardiovascular pathology in mucopolysaccharidosis I mice.Lice Science. 2018;196:102-109.
3. Haskins ME. Animal models of mucopolysaccharidosis and their clinical relevance. Acta Paediatr. 2007;96:56-92.
4. Haskins ME, Aguirre GD, Jezyk PF, Desnick RJ, Patterson DF. The pathology of the feline model of mucopolysaccharidosis I. Am J Pathol. 1983:112:27-36.
5. Hinderer C, Katz N, Louboutin J-P, et al. Abnormal polyamine metabolism is unique to the neuropathic forms of MPS: potential for biomarker development and insight into pathogenesis. Human Molec Genet. 2017; 26:3837-3849.
6. Khalid O, Vera MU, Gordts PL, et al. Immune-mediated inflammation may contribute to the pathogenesis of cardiovascular disease in mucopolysaccharidosis. Plos One. 2016;DOI:10.1371/journal.pone.010850.
7. Lyons JA, Dickson PI, Wall JS, et al. Arterial pathology in canine mucopolysaccharidosis-I and response to therapy. Lab Invest. 2010;91:665-674.
8. Menon KP, Tieu PT, Neufeld EF. Architecture of the canine IDUA gene and mutation underlying canine mucopolysaccharidosis I. Genomics 1992;14:763-768.
dosis I. Genomics 1992; 14:763-768.
9. Neufeld E, Muenzer JA. The mucopolysaccharidoses. Ch 61 in
Eds. Scriver CR, Beaudet AL, Sly WS,Valle D. The Metabolic Basis of Inherited Disease, vol 2, 6th ed. UDA:McGraw-Hill; 1989:1565-1587.
10. Parini R, Deodoro F, Di Rocco M, et al. Open issues in mucopolysaccharidosis I-Hurler. Orphanet J Rare Dis. 2017;12:112. DOI 10.1186/s13023-017-0662-9.
11. Shull RM, Helman RG, Spellacy E, Constantopoulos G. Munger RJ, Neufeld EF. Morphologic and biochemical studies of canine mucopolysaccharidosis I. Am J Pathol. 1984;114:487-495.
12. Spellacy E, Shull RM, Constantopoulos G, Neufeld EF. A canine model of human alpha-L-iduronidase deficiency. Proc Natl Acad Sci USA. 1983;80:6091-3065.