Signalment:
Gross Description:
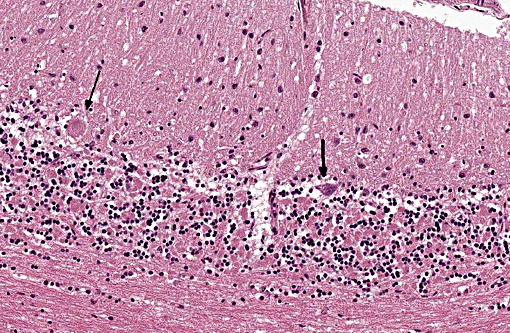
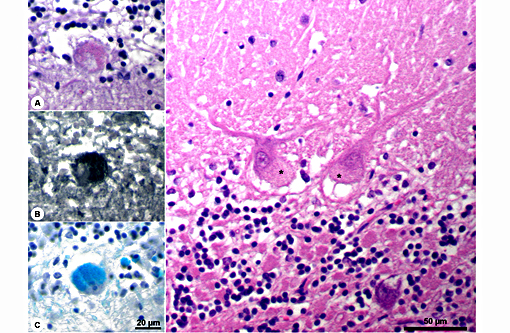
Histopathologic Description:
Morphologic Diagnosis:
Condition:
Contributor Comment:
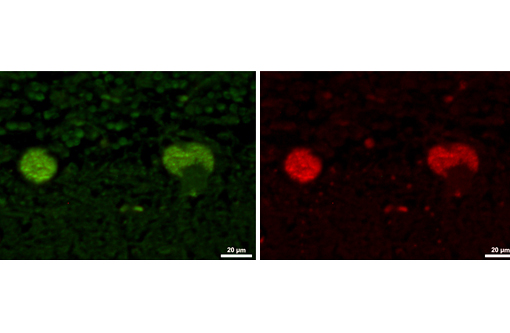
The disease is characterized by accumulation of granules in the cytoplasm of most nerve cells and, to a lesser extent, of many other cell types. The granules are autofluorescent, periodic acid-Schiff (PAS), Luxol Fast Blue and Sudan black B positive and resistant to lipid solvents. Consequences include progressive selective neuronal loss with secondary astrocytic proliferation and hypertrophy as well as infiltration by macrophages.
The disease is clinically classified by age of onset into early (up to 1.5 years) and late (up to 9 years) onset forms. It is additionally categorized according to the defective genes associated with the disease (see Table 1). All forms result in progressive neurodegen-eration of the central nervous system, but clinical symptoms are variable depending on breed, age and individual factors. Affected dogs change their behavior, become blind, and suffer from progressive proprioceptive and motor deficits, seizures and ataxia.
Gross lesions are often absent; especially in cases with early onset, marked cerebral atrophy may be apparent with often a brown tinge in severely affected areas.
JPC Diagnosis:
Conference Comment:
Storage diseases comprise a heterogeneous group of conditions that result from the accumulation of material within the cell, which is unable to be metabolized or further broken down, either due to resistance to cellular processing or excessive accumulation that exceed the cells capacity to process it. Many of the storage diseases involve lysosomes, and in animals nearly all of the inherited storage diseases are lysosomal in nature. They often affect neurons due to the long-lived nature of these cells, which provides a longer period over which material can be accumulated as compared to cells with short turnover times.
Defects in lysosomal processing may involve defects in a specific enzyme or lysosomal hydrolase due to a genetic defect, and can include defects in post-translational processing, production, or trafficking. Non-inherited mechanisms of storage disease include ingestion of an exogenous toxin which inhibits a lysosomal enzyme, with the result being similar to those involving genetic defects. This is the case with ingestion of locoweed which produces the phytotoxin swainsonine. Resistance of the accumulating material to cellular processing may also be due to specific alterations in the substrate, either exogenous or endogenous, and not due to a processing enzyme defect per se. The detailed mechanisms involved in cellular dysfunction and death, secondary to the accumulation of undigested material, in most cases are unclear.(5)
| Breed | Reference | Gene | Onset | Age |
| American Bulldog | 4,34 | CTSD | Early | 1 3 Years |
| American Staffordshire Terrier | 1 | ARSG | Late | 1.5 9 Years |
| Australian Cattle Dog | 28 | Early | 12 Month | |
| Australian Shepherd | 17,23 | CLN6 | Early | 18 21 Month |
| Border Collie | 19 | CLN5 | Early | 15 Month |
| Chihuahua | 21 | Early | 1 2 Years | |
| Chinese Crested dog | 12 | MFSD8 | Early | 19 Month |
| Cocker Spaniel | 20 | Late | 1.5 6 Years | |
| Dachshund | 3,27 | TPP1 (CLN2) | Early | 6 12 Month |
| Dachshund | 26 | CLN1 | Early | 9 Month |
| Dachshund | 32 | Late | 4.5 7 Years | |
| Dalmatian | 9,10 | Early | 6 12 Month | |
| English Setter | 18 | CLN8 | Early | 1 1.5 Years |
| Labrador Retriever | 25 | Late | 7 Years | |
| Miniature Schnauzer | 15,24 | Late | 2 4 Years | |
| Mixed breed (Australian Shepherd / Blue Heeler) | 11 | CLN8 | Early | 8 Month |
| Polski Owczarek Nizinny (PON) | 22 | Early-late | 0.5 4.5 Years | |
| Retriever | 30 | Late | 3 Years | |
| Saluki | 2 | Early | 12 Month | |
| Tibetan Terrier | 6,35 | ATP13A2 | Late | 4 8 Years |
| Welsh Corgi | 14 | Late | 6 8 Years |
In the veterinary literature neuronal ceroid-lipofuscinoses (NCL) is best characterized in sheep and dogs, and as described in the above chart, the condition in dogs is generally classified by age of onset and course of disease.(21) The actual defect resulting in abnormal accumulations may not only involve lysosomal enzymes, but also mitochondrial defects or other defects in protein processing involving the endoplasmic reticulum. The variation in gene mutations and resulting defects likely play a role in the variation in clinical presentations and histologic findings. The storage material seen within the Purkinje cell cytoplasm can be present in multiple organs, but is particularly damaging in neurons, and may be seen in the cerebral cortical and retinal neurons in addition to Purkinje cells. Severe blindness may be the primary clinical presentation in which case retinal atrophy would likely be the primary lesion, whereas other animals will present with gait abnormalities, aggression, seizures or dementia with loss of Purkinje cells and/or neurons within other regions of the brain (hippocampus, cerebrum, brainstem).(5) Lipofuscin accumulates in many cells throughout the body as part of the normal aging process, and in the retina is normally only seen in the retina pigment epithelium. Autoflourescent material within other retinal cells, including ganglion and Mueller cells, is diagnostic for neuronal ceroid lipofuscinosis.(11)
References:
1. Abitbol M, Thibaud J-L, Olby NJ, Hitte C, Puech J-P, et al. A canine Arylsulfatase G (ARSG) mutation leading to a sulfatase deficiency is associated with neuronal ceroid lipofuscinosis. PNAS. 2010;107:14775-14780.
2. Appleby EC, Longstaffe JA, Bell FR. Ceroid-lipofuscinosis in two Saluki dogs. J Comp Path. 1982; 92: 375-380.
3. Awano T, Katz ML, O'Brien DP, Sohar I, et al. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Molec Genet Metab. 2006; 89: 254-260.
4. Awano T, Katz ML, O'Brien DP, Taylor JF, et al. A mutation in the cathepsin D gene (CTSD) in American bulldogs with neuronal ceroid lipofuscinosis. Molec Genet Metab. 2006; 87: 341-348.
5. Cantile C, Youssef S. Nervous System. In: Maxie MG, ed. Jubb, Kennedy, and Palmer's Pathology of Domestic Animals. Vol 1. 6th ed. St. Louis, MO: Elsevier; 2015:284-292.
6. Farias FHG, Zeng R, Johnson GS, Wininger FA, et al. A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiology of Disease. 2011; 42: 468-474.
7. Fiske RA, Storts RW. Neuronal ceroid-lipofuscinosis in Nubian goats. Vet Pathol. 1988; 25: 171-173.
8. Gao HL, Boustany RMN, Espinola JA, Cotman SL, et al. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet. 2002; 70: 324-335.
9. Goebel HH, Bilzer T, Dahme E, Malkusch F. Morphological studies in canine (Dalmatian) neuronal ceroid-lipofuscinosis. Am J Med Genet Suppl. 1988; 5: 127-139.
10. Goebel HH, Dahme E. Ultrastructure of retinal pigment epithelial and neural cells in the neuronal ceroid-lipofuscinosis affected Dalmatian dog. Retina. 1986; 6: 179-187.
11. Guo J, Johnson GS, Brown HA, Provencher ML. A CLN8 nonsense mutation in the whole genome sequence of a mixed breed dog with neuronal ceroid lipofuscinosis and Australian shepherd ancestry. Molec Genet Metab. 2014; 112: 302-309.
12. Guo JY, O'Brien DP, Mhlanga-Mutangadura T, Olby NJ. A rare homozygous MFSD8 single-base-pair deletion and frameshift in the whole genome sequence of a Chinese crested dog with neuronal ceroid lipofuscinosis. BMC Vet Res. 2015;10:960.
13. Harper PAW, Walker KH, Healy PJ, Hartley WJ, et al. Neurovisceral ceroid-lipofuscinosis in blind Devon cattle. Acta Neuropathologica. 1988; 75: 632-636.
14. Jolly RD, Palmer DN, Studdert VP, Sutton RH, et al. Canine ceroid-lipofuscinoses: A review and classification. Journal of Small Animal Practice. 1994; 35: 299-306.
15. Jolly RD, Sutton RH, Smith RIE, Palmer DN. Ceroid-lipofuscinosis in miniature schnauzer dogs. Aust Vet J. 1997; 75: 67-67.
16. Karli P, Karol A, Oevermann A. The canine neuronal ceroid-lipofuscinosis: A review. Schweizer Archiv Fur Tierheilkunde. 2014;156: 417-423.
17. Katz ML, Farias FH, Sanders DN, Zeng R. A missense mutation in canine CLN6 in an Australian shepherd with neuronal ceroid lipofuscinosis. J Biomed Biotechnol. 2011; 2011:198042.
18. Katz ML, Khan S, Awano T, Shahid SA, et al. A mutation in the CLN8 gene in English Setter dogs with neuronal ceroid-lipofuscinosis. Biochem Biophys Res Commun. 2005; 327: 541-547.
19. Melville SA, Wilson CL, Chiang CS, Studdert VP. A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in border collie dogs. Genomics. 2005; 86: 287-294.
20. Minatel L, Underwood SC, Carfagnini JC. Ceroid-lipofuscinosis in a Cocker spaniel dog. Vet Pathol. 2000; 37: 488-490.
21. Nakamoto Y, Yamato O, Uchida K, Nibe K. Neuronal Ceroid-Lipofuscinosis in longhaired chihuahuas: Clinical, pathologic, and MRI findings. J Am Anim Hosp Assoc. 2011; 47: E64-E70.
22. Narfstrom K, Wrigstad A, Ekesten B, Berg AL. Neuronal ceroid lipofuscinosis: clinical and morphologic findings in nine affected Polish Owczarek Nizinny (PON) dogs. Vet Ophthalmol. 2007; 10: 111-120.
23. O'Brien DP, Katz ML. Neuronal ceroid lipofuscinosis in 3 australian shepherd littermates. J Vet Intern Med. 2008; 22: 472-475.
24. Palmer DN, Tyynela J, vanMil HC, Westlake VJ, Jolly RD. Accumulation of sphingolipid activator proteins (SAPs) A and D in granular osmiophilic deposits in miniature schnauzer dogs with ceroid-lipofuscinosis. J Inherit Metab Dis. 1997; 20: 74-84.
25. Rossmeisl JH, Duncan R, Fox J, Herring ES, Inzana KD. Neuronal ceroid-lipofuscinosis in a Labrador retriever. J Vet Diag Invest. 2003; 15: 457-460.
26. Sanders DN, Farias FH, Johnson GS, Chiang V. A mutation in canine PPT1 causes early onset neuronal ceroid lipofuscinosis in a dachshund. Molec Genet Metab. 2010; 100: 349-356.
27. Sanders DN, Kanazono S, Wininger FA, Whiting REH. A reversal learning task detects cognitive deficits in a dachshund model of late-infantile neuronal ceroid lipofuscinosis. Genes Brain and Behavior. 2011; 10: 798-804.
28. Sisk DB, Levesque DC, Wood PA, Styer EL. Clinical and pathologic features of ceroid lipofuscinosis in two Australian cattle dogs. J Am Med Assoc. 1990; 197: 361-364.
29. Tammen I, Houweling PJ, Frugier T, Mitchell NL, et al. A missense mutation (c. 184C > T) in ovine CLN6 causes neuronal ceroid lipofuscinosis in Merino sheep whereas affected South Hampshire sheep have reduced levels of CLN6 mRNA. Biochim Biophys Acta. 2006;1762: 898-905.
30. Umemura T, Sato H, Goryo M, Itakura C. Generalized lipofuscinosis in a dog. Japanese J Vet Sci. 1985; 47: 673-677.
31. Url A, Bauder B, Thalhammer J, Nowotny N, et al. Equine neuronal ceroid lipofuscinosis. Acta Neuropathologica. 2001; 101: 410-414.
32. Vandevelde M, Kristensen B, Braund KG, Greene CE. Chronic canine distemper virus encephalitis in mature dogs. Vet Pathol. 1980; 17: 17-28.
33. Weissenboeck H, Rossel C. Neuronal ceroid-lipofuscinosis in a domestic cat: Clinical, morphological and immunohistochemical findings. J Comp Path. 1997; 117: 17-24.
34. Woehlke A, Droegemueller C, Distl O. Canine ceroid lipofuscinosis in American bulldogs. Tieraerztliche Praxis Ausgabe Kleintiere Heimtiere. 2007; 35: 351.
35. Woehlke A, Philipp U, Bock P, Beineke A, et al. A one base pair deletion in the canine ATP13A2 gene causes exon skipping and late-onset neuronal ceroid lipofuscinosis in the Tibetan terrier. PLoS Genet. 2011; 7(10): e1002304.