CASE III: S18-1972 (JPC 4136167)
Signalment:
10-year-old, female, Chihuahua, dog, Canis lupus familiaris
History:
History of coughing, suspicion of aspiration pneumonia and megaesophagus. Congenital heart malformation (Ventricular Septal Defect = VSD) without signs of decompensation. Epileptic attacks since three years, under treatment with levetiracetam, imepitoin and phenobarbital.
Gross Pathology:
At necropsy, the dog was in a good body condition with ample body fat. The left cranial lobe of the lung was firm and had a red color. The remaining lung tissue had a red color and was of normal consistency. The macroscopic examination of the heart confirmed the presence of a VSD, which had been previously diagnosed by echocardiography, and measured approximately 1 cm in diameter. Pronounced right atrial myocardial dilation was present mostly likely resulting from a chronic volume overload due to the VSD. The weight of the heart was 44g (relative heart weight: 1.76%, Reference value in dogs: 0.5-1%). Other findings included a moderate dilation of the cranial and middle part of the esophagus. All other organs, in particular the brain, were unremarkable during macroscopic examination.
Laboratory results:
No laboratory findings reported.
Microscopic Description:
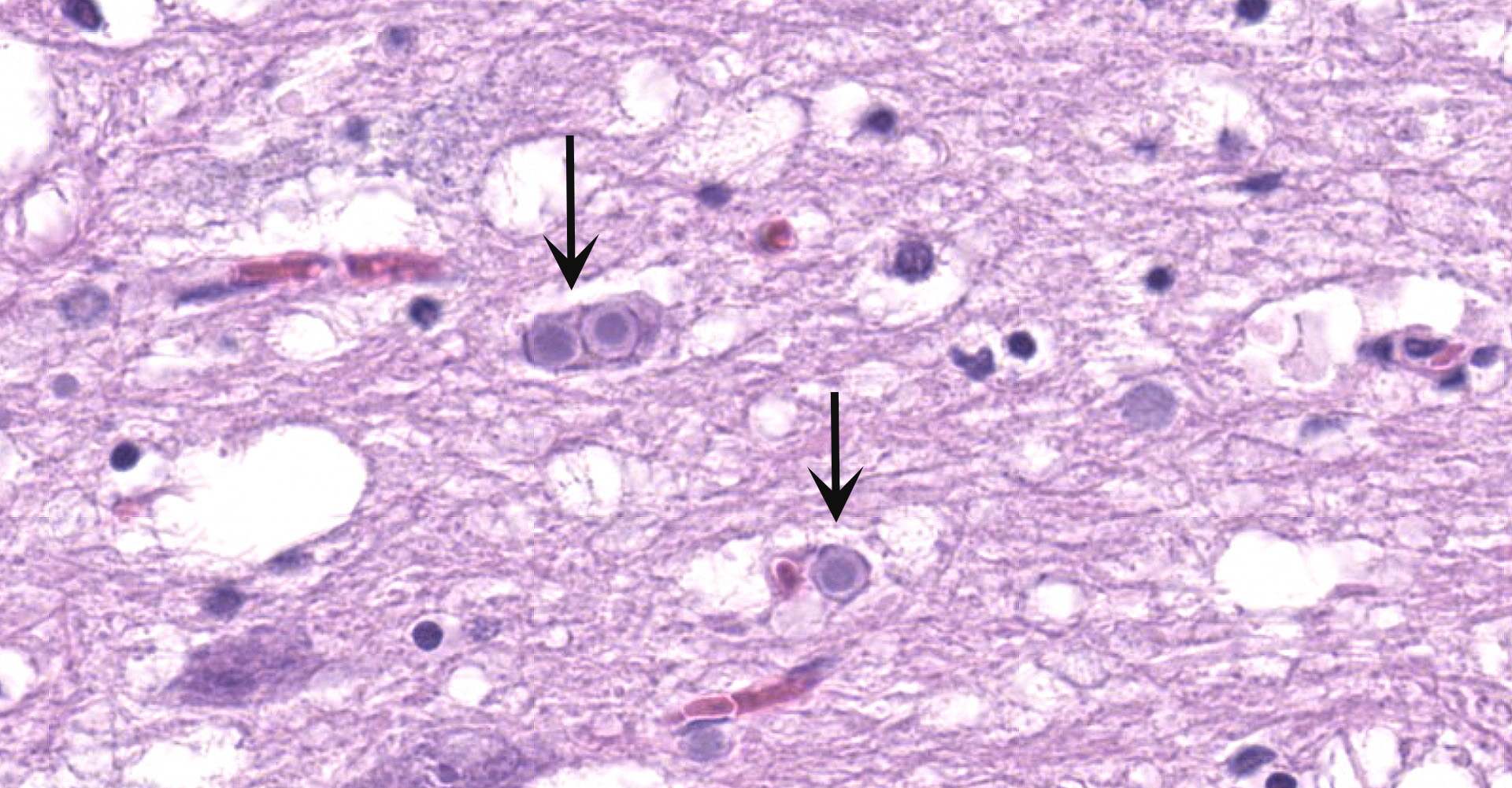
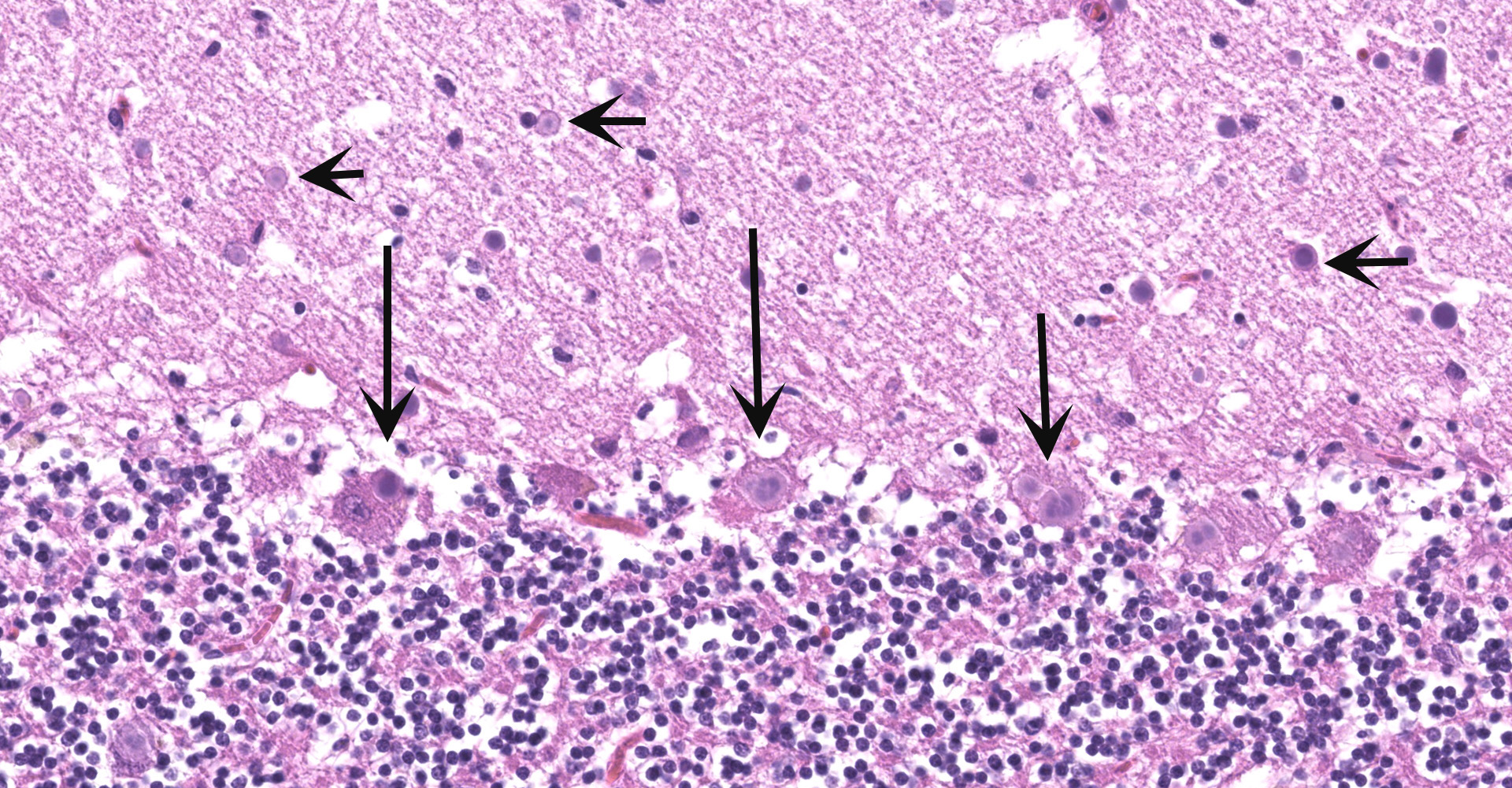
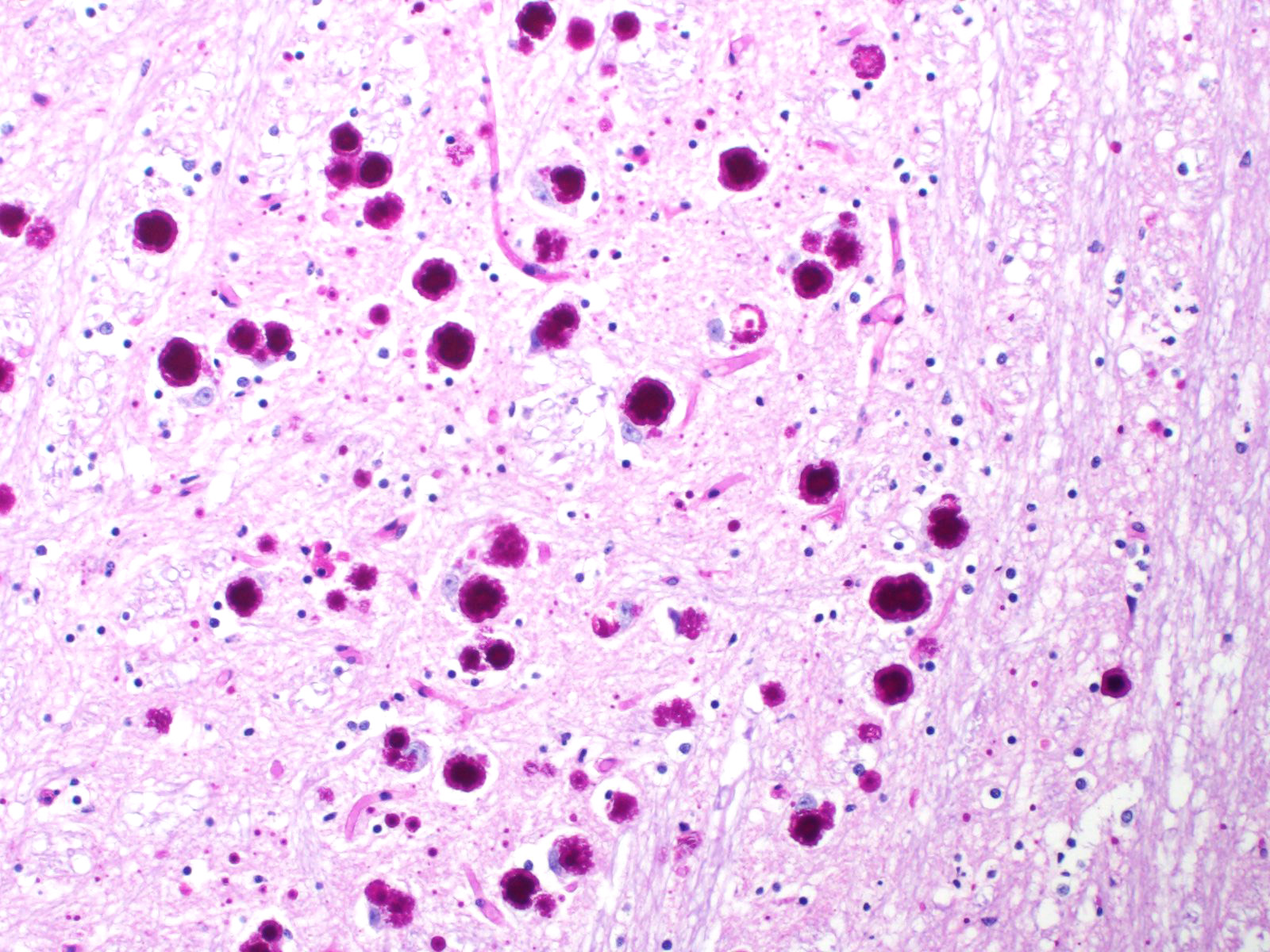
Cerebellum and brainstem: Multifocal and randomly distributed, there are few to many lightly basophilic inclusions within the white and grey matter, as well as in all layers of the cerebellum, the neuronal perikarya, neuronal processes and scattered within the neuropil. The inclusions are round to globular, range from 2 to 10 µm in diameter and have occasionally a pale red center. The peripheral margins are either smooth or have a faint, perpendicularly oriented striation and most deposits have a thin clear halo. Additionally, there is mild, multifocal satellitosis and mild diffuse increase in microglia.
Contributor's Morphologic Diagnoses:
Cerebellum and brainstem: numerous intraneuronal basophilic inclusions (polyglucosan bodies); mild multifocal satellitosis and microgliosi.
Contributor's Comment:
Significant histologic lesions of the brain involved the cerebrum, the cerebellum, the brainstem including the nuclei of the vagus nerve.
All basophilic inclusions were strongly Periodic Acid-Schiff (PAS) positive and were therefore identified as polyglucosan or Lafora-like bodies. A total of nine histologic sections of brain and vagus nerve were examined.
No other histologic changes other than the presence of these polyglucosan deposits were identified in the brain and vagus nerve. The polyglucosan or Lafora-like bodies were most numerous in the molecular layer of the cerebellum and in the brainstem, smaller numbers were seen in other brain localizations or in the vagus nerve. No deposits were identified in other tissues such as liver, cardiac and skeletal muscle or esophagus.
The histological examination of the lung within the left cranial lobe revealed that the alveoli were filled with a large number of intact or degenerated neutrophils, accumulations of fine fibrillar eosinophilic material (fibrin) and protein-rich edema fluid and occasional cellular debris (necrosis). Within the alveoli, fine granular, green material was also detected, and interpreted as aspiration of foreign material, resulting from the CNS symptoms. Alveolar septa were moderately congested and frequently necrotic. Interlobular septa were markedly expanded/distended by fibrin, edema fluid, and inflammatory cells (mostly neutrophils).
The histological examination of the heart was unremarkable.
Lafora disease (LD) is an autosomal recessive disorder that causes non-fatal myoclonic epilepsy.2,6 The disease is characterized by the presence of polyglucosan inclusion bodies (Lafora bodies), predominantly in the central nervous system.
More than 90% of human LD cases are caused by genetic variants in one of two different genes, EPM2A, encoding laforin glucan phosphatase, or EPM2B (also known as NHLRC1) encoding the NHL repeat containing E3 ubiquitin protein ligase 1, also termed malin.10,11,12
LD in animals has similar clinical signs as the human disease, including spontaneous and reflex myoclonus, jerks and generalized tonic clonic seizures.
In animals, LD has been reported in the dog, cat, cow, and fennec fox.7,8,9 In dogs, LD is one of the most commonly recognized structural-metabolic epilepsies. A genetic abnormality has been identified only in canine LD, in association with a repeat expansion in the EPM2B gene11 causing a functional impairment of the ubiquitin ligase malin, which regulates the glycogen metabolism. Thus, abnormally structured glycogen accumulates and forms polyglucosan bodies, predominantly in the nervous system.
Miniature wirehaired dachshund is the breed most commonly affected. LD cases with EPM2B repeat expansion have also been reported in the basset hound and beagle. 7,11 The diagnosis of LD is based on functional (clinical sign), genotypic (gene mutation), and phenotypic (histopathology) examinations. Histopathologically, LD is characterized by the presence of pathognomonic inclusions named Lafora bodies especially in the brain, spinal cord and other tissues such as skin, liver, cardiac and skeletal muscle.
In this case, no further genetic examination was undertaken.
Contributing Institution:
Institute of Veterinary Pathology, Vetsuisse Faculty (University of Zurich),
Winterthurerstrasse 268, CH-8057 Zurich, Fax number +41 44 635 89 34
JPC Diagnosis:
Brainstem and cerebellum: Intraneuronal Lafora (polyglucosan) bodies, numerous, with mild scattered microgliosis and spongiosis.
JPC Comment:
Lafora disease bears the name of Gonzalo Rodríguez-Lafora, who discovered the characteristic intraneuronal bodies in 1911. Born in Madrid in 1886 and the son of a Spanish army officer, Lafora spent part of his childhood in the Spanish colony of Puerto Rico (before the Spanish-American War of 1898) before attending medical school at the Central University of Madrid and receiving his medical degree at the age of 21. He went on to pursue additional study in the field of neuropsychiatry at multiple prominent Spanish, French, and German institutions in the years that followed. In 1910, he was appointed as the neuropathologist for the Government Hospital for the Insane in Washington D.C., during which time he discovered the intraneuronal bodies that bear his name today while studying familial myoclonic epilepsy. Throughout his career, Lafora published approximately 200 papers, including original works on sleep, neurosyphilis, histological alterations in dementias, and studies on schizphrenia. In addition to his professional life as a neurologist, pathologist, and psychiatrist, Lafora was also an impressionist painter, specializing in cubism.13
Interestingly, the first clinical signs of Lafora disease in both humans and dogs occur at a similar chronological age (in dogs at seven years of age and during late childhood/adolescence in humans). The disease then progresses at a similar rate of the remaining lifetime of the dog and over 10 years in humans. This is unique in that other equivalent neurodegenerative diseases between the two species occur at the same epigenetic age (i.e. a disease affecting an adolescent human will occur in an 8-10 month old puppy).16
As noted by the contributor, Lafora disease has no cure and is one of the severest forms of familial myoclonic epilepsies observed in several species, but particularly in humans and dogs. In dogs, Lafora disease is caused by a repeat expansion in the NHL repeat containing the E3 ubiquitin protein ligase 1 (NHLRC1) gene. Compared to other species, dogs are predisposed to Lafora disease, with affected individuals carrying 19-26 copies of the repeat sequence rather than the expected 2-3.16
Under normal conditions, glucose is stored as soluble glycogen. However, in the absence of NHLRC1 (EPM2B), glycogen is malstructured and becomes an insoluble complex polyglucosan.3,16 Over time, this abnormal glycogen accumulates into Lafora bodies in neurons and astrocytes. This results in progressive neuronal degeneration, which is driven by impaired astrocytic function and neuroinflammation. The mechanism in which Lafora disease results in myoclonic epilepsy is uncertain. However, there is increasing evidence that accumulation of glycogen within astrocytes plays a central role in the pathogenesis. It is hypothesized that inflammation, astrocyte reactivity, and activation of microglia play a major role in the triggering of seizures. Interestingly, blocking brain glycogen synthesis in mouse Lafora disease models prevents disease progression.16
In humans, Lafora disease clinically manifests as myoclonus (photomyoclonic response), tonic-clinic seizures, visual hallucinations, and blindness. The disease rapidly progresses, leading to more severe and frequent seizures with increased refractoriness, ataxia, dementia, and eventually a vegetative state. Human patients typically die within 10 years of onset due to status epilepticus. Dogs demonstrate a similar clinical progression, with the addition of aggression and incontinence, and are typically euthanized when quality of life is significantly impacted by the disease.14,16
Although Lafora bodies are a characteristic feature of Lafora disease, the presence of these 5-20 µm, periodic acid-Schiff (PAS) positive, basophilic, intracytoplasmic Lafora bodies is not pathognomonic for the disease. In fact, Lafora bodies are most frequently identified as incidental findings in aged animals. However, in cases of Lafora disease they occur in massive numbers, such as in this case.3
Lafora disease is an autosomal recessive species wide disorder in canines that will likely continue to spontaneously appear in many breeds and mixed breed dogs. Given this disease has been reported in multiple breeds, genetic testing should be considered when designing breeding strategies in affected breeds, especially those with small genetic pools. This is complicated by the fact Lafora disease has a late onset in dogs and many are bred before developing clinical signs. Muscle biopsy is a useful ancillary test, especially for breeds where genetic tests are available or have not been sufficiently validated. The optimal site for muscle biopsy is the tissue adjacent to the myotendinous insertion and the myofascial union as Lafora bodies are only sparsely scattered in the muscle belly area.16 Interestingly, a 2011 study evaluating quadriceps biopsies from mice identified Lafora bodies only within type II myofibers, with none found within type I fibers.15 This finding has since been documented within canine skeletal muscle.4
In addition to the mainstay treatment of antiepileptic medications, ketogenic diets have been receiving attention in regard to management of this disease. These diets maintain blood glucose concentrations in the low normal range, resulting in the brain using ketones for energy production. In addition, this type of diet is also antiepileptic due to γ-glutamyl transpeptidase and γ-glutamylation inhibition. A lifelong ketogenic diet in a mouse model of Lafora disease reduced the number of Lafora bodies; however, human studies were less promising but did suggest the disease's progression was delayed if a ketogenic diet was started early in the process. Ketogenic diets have not yet been trialed for dogs with Lafora disease, although their utilization is often used on an empirical basis.16
Determination of the morphologic diagnosis elicited spirited and robust discussion amongst participants. Approximately half favored "degenerative encephalopathy" as the primary lesion being due to the presence of insoluble Lafora bodies within neurons, an inherently degenerative lesion ultimately resulting in neuronal dysfunction. Remaining participants favored the morphologic diagnosis of "intraneuronal Lafora bodies" based on the paucity of neurons exhibiting classic characteristics of degeneration, including cell shrinkage, loss of Nissl substance, intensely eosinophilic cytoplasm, and a pyknotic nucleus. Regardless of institutional and training background, there is no debate in regard to the degenerative and progressive nature of Lafora disease as an entity.
References:
1. Andrade DM, Turnbull J, Minassian BA. Lafora disease, seizures and sugars. Acta Myol. 2007;26:83-86.
2. Berendt M, Farquhar RG, Mandigers PJ, et al. International veterinary epilepsy task force consensus report on epilepsy definition, classification and terminology in companion animals. BMC Vet. Res. 2015;11:182.
3. Cantile C, Youssef S. Nervous system. In: Maxie MG, ed. Jubb, Kennedy, and Palmer's Pathology of Domestic Animals. Vol 1. 6th ed. Philadelphia, PA: Elsevier Saunders; 2016: 255, 292.
4. Chambers JK, Thongtharb A, Shiga T, et al. Accumulation of Laforin and Other Related Proteins in Canine Lafora Disease With EPM2B Repeat Expansion. Vet Pathol. 2018;55(4):543-551.
5. Chan EM, Young EJ, Ianzano L, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35:125-127.
6. Delgado-Escueta AV. Advances in Lafora progressive myoclonus epilepsy. Curr Neurol Neurosci Rep. 2007;7(5):428-433.
7. Hajek I, Kettner F, Simerdova V, et al. NHLRC1 repeat expansion in two beagles with Lafora disease. J Small Anim Pract. 2016;57(11):650-652.
8. Hall DG, Steffens WL, Lassiter L. Lafora bodies associated with neurologic sign in a cat. Vet Pathol. 1998;35(3):218-220.
9. Honnold SP, Schulman FY, Bauman K, Nelson K. Lafora's-like disease in a fennec fox (Vulpes zerda). J Zoo Wildl Med. 2010;41(3):530-534.
10. Ianzano L. et al. Lafora progressive myoclonus epilepsy mutation database‐EPM2A and NHLRC1 (EMP2B) genes. Human Mutat. 2005;26:397.
11. Lohi H, Young EJ, Fitzmaurice SN, et al. Expanded repeat in canine epilepsy. Science. 2005;307(5706):81.
12. Minassian BA, Ianzano L, Delgado-Escueta AV, et al. Identification of new and common mutations in the EPM2A gene in Lafora disease. Neurology. 2000;54:488-490.
13. Nanduri AS, Kaushal N, Clusmann H, Binder DK. The maestro don Gonzalo Rodríguez-Lafora. Epilepsia. 2008;49(6):943-947.
14. Swain L, Key G, Tauro A, Ahonen S, Wang P, Ackerley C, Minassian BA, Rusbridge C. Lafora disease in miniature Wirehaired Dachshunds. PLoS One. 2017 Aug 2;12(8):e0182024.
15. Turnbull J, Girard JM, Pencea N, et al. Lafora bodies in skeletal muscle are fiber type specific. Neurology. 2011;76(19): 1674-1676.
16. von Klopmann T, Ahonen S, Espadas-Santiuste I, et al. Canine Lafora Disease: An Unstable Repeat Expansion Disorder. Life (Basel). 2021;11(7):689. Published 2021 Jul 14.