Signalment:
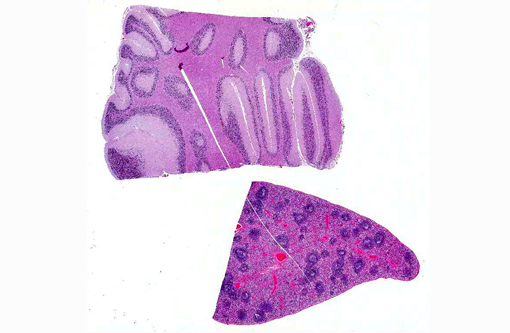
Gross Description:
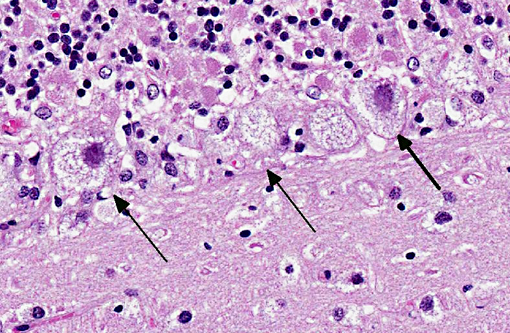
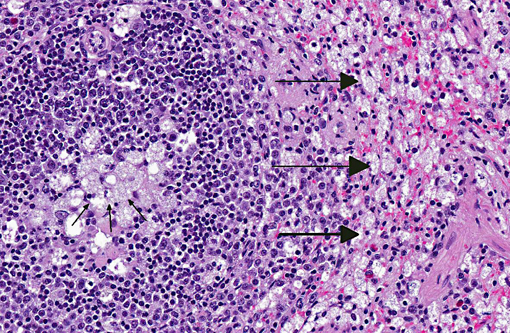
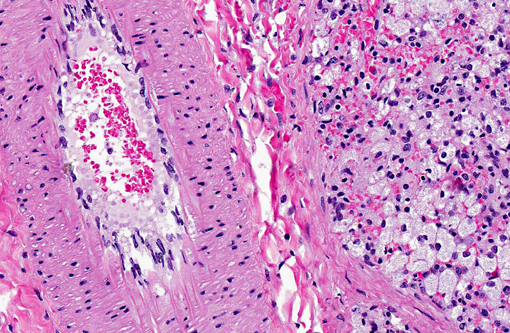
Histopathologic Description:
Morphologic Diagnosis:
Cerebellum: Severe diffuse neuronal and glial cell vacuolation and swelling with occasional multifocal spheroids (suspect storage disease).
Spleen: Germinal centers and red pulp severe diffuse histiocytosis (suspect storage disease).
Lab Results:
| Test | Result |
| Aerobic culture- lung, liver, mesenteric lymph node | Mixed growth |
| Fecal PCR for Salmonella sp. | Salmonella arizonae |
| Fecal flotation | Negative |
| Serology for Toxoplasma sp. | Negative |
| Heavy/trace mineral analysis (lead, manganese, iron, zinc, arsenic, cadmium, molybdenum, copper, mercury, selenium) | Within normal limits |
| Rabies testing via fluorescent antibody testing on brain tissue | Negative |
| Lysosomal enzyme analysis | See Table 1 |
| Special stains: Oil red O (on formalin fixed frozen sections), PAS, Sudan Black, Luxol fast blue and acid fast | - Multifocal endothelial cell intra-cytoplasmic accumulation of Oil red O-positive material - Affected neurons and macrophages were Oil red O, PAS, Sudan Black, Luxol fast blue and acid fast negative |
| Autofluorescence (via UV scope) | Negative |
Condition:
Contributor Comment:
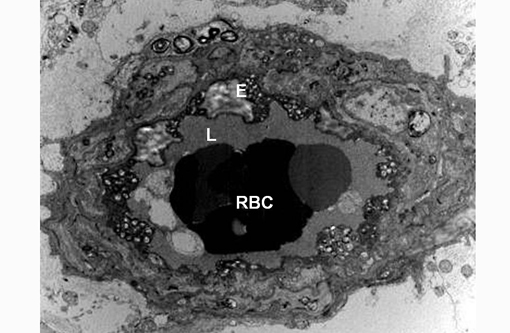
Transmission electron microscopy revealed lysosomal accumulations of floccular variably electron dense and frequently concentrically arranged lamellar material consistent with lysosomal storage disease. However, ultrastructural analysis is relatively non-specific regarding type of storage.
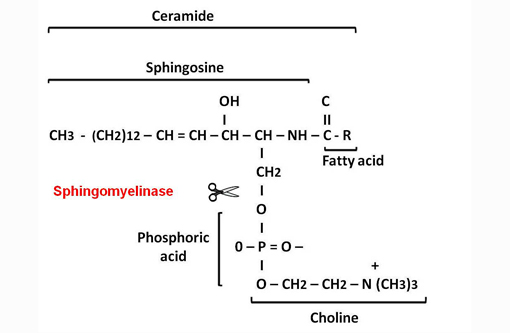
Measurement of lysosomal enzyme activity including sphingomyelinase, β-galactosidase, β-hexosaminidase and β-hexosaminidase A and B was performed in water homogenates of the brain samples from affected and age-matched non-affected raccoon(8) (Table 1). This assay revealed complete absence of sphingomyelinase activity. The absence of sphingomyelinase activity is a criterion for the diagnosis of sphingomyelin lipidosis (also known as Niemann Pick disease (NPD)).(7)
Sphingomyelin lipidosis belongs to a large group of sphingolipidosis lysosomal storage diseases that also includes GM1 and GM2 gangliosidosis and globoid cell leukodystrophy, to name a few. In sphingolipidoses the spectrum of affected organs is wide and often includes viscera and macrophages because the substrate is derived from all cell membranes.(3) The involvement of neurons in most LSDs is due to both the high metabolic activity of these cells and their long life span, which allows the gradual accumulation of undegraded substrate.(3) Ultrastructural pathology offers useful information in the diagnosis of LSDs and helps categorize the type of LSD. In diseases accumulating sphingolipids, storage bodies are characterized by membranous material arranged concentrically (membranous cytoplasmic bodies). None of these forms is specific for a given disease, but concentric lamellae are most common in GM1 and GM2 gangliosidoses. In the present case the storage bodies were poorly defined, concentrically arranged lamellar whorls. Histochemistry, immunohistochemistry and fluorescence microscopy may also be of use in identifying storage material, but definitive and gold standard for diagnosis is by means of biochemical analysis.(3)
There is no single presentation common to all lysosomal storage diseases; the clinical and gross pathological manifestations are dependent on the deficient enzyme and the outcome of the deficiency in the organs that utilize the enzyme. Microscopically, LSDs are characterized by accumulation of enlarged lysosomes containing uncatabolized substrate in solution or complexed with related chemical species, which will be partially or wholly removed during fixation and preparation of the paraffin wax-embedded sections. If the substrate is soluble in water or lipid solvents, there will be a vacuolated appearance of the affected cells.(3)
Sphingolipids are an important group of structural lipids in which the unifying compound, ceramide, is esterified to sialyloligosaccharides to form gangliosides, to other saccharides to form neutral glycolipids such as globoside, or to phosphocholine to form sphingomyelin.(3)
The primary metabolic defect in NiemannPick disease (NPD) types A and B in man is the lack of sphingomyelinase enzyme that catalyzes the hydrolytic cleavage of sphingomyelin to ceramide and phosphocholine.(3) The reduced or absent enzyme activity results in accumulation of sphingomyelin in lysosomes.(7) Similarly to humans, an autosomal recessive mode of inheritance has been demonstrated in a cat and a dog.(2,8)
Similar histological and ultrastructural features to those in this case were reported affecting neurons, oligodendroglial cells, macrophages, renal epithelial cells, endothelium and pericytes in the CNS, PNS, spleen, liver, lung and kidney in a cat,(1) a dog and a Hereford calf.(6) Consistent with the present case, these animals presented as juveniles, had neurological signs and virtually no sphingomyelinase activity was detected in the brain and liver compared with normal controls. Autofluorescence by UV light was reported in a dog with NPD,(2) but was not found in the present case.
Table 1: Lysosomal enzyme activities in brain homogenates from the affected and a normal raccoon
| Enzyme activity (nmol/h/mg protein) | Affected Raccoon | Normal Raccoon |
| Beta-galactosidasea | 64.0 | 22.6 |
| Beta-hexosaminidase Ab | 81.4 | 28.9 |
| Beta-hexosaminidase A and Bc | 611.1 | 118.0 |
| Sphingomyelinased | 0.13 (3X) | 5.2 |
b- measured using 4MU-β-N-acetylglucosaminideSO4
c- measured using 4MU-β-N-acetylglucosaminide
d- measured using 14C-sphingomyelin; 3x means measured three times
JPC Diagnosis:
Cerebellum: Neuronal, glial cell and endothelial cytoplasmic vacuolation, diffuse, marked, with gliosis.
Spleen: Histiocytosis, diffuse, marked with cytoplasmic vacuolation.
Conference Comment:
Globoid cell leukodystrophy, also known as galactocerebrosidosis, is an autosomal recessive disorder reported in dogs, cats and polled Dorset sheep. It is classified within the sphingolipidosis group of storage diseases and results from deficient activity lysosomal galactocerebrosidase, an enzyme which normally catalyzes the breakdown of galactocerebrosides. Galactocerebrosides are important components of myelin, but at high concentrations are cytotoxic; excessive accumulation in oligodendrocytes and Schwann cells causes extensive cellular degeneration and necrosis, halting active myelination. This combined with the degeneration of existing myelin, results in demyelination and axonal loss. Phagocytic macrophages, however, are unable to degrade galactocerebroside, and thus appear microscopically as characteristic swollen, PAS-positive globoid cells, which exhibit perivascular cuffing within the white matter. In contrast to many of the other lysosomal storage diseases, neurons are not typically involved in the accumulation of excess storage material in galactocerebrosidosis.(5)
Glycoproteinoses, such as α- and β-mannosidosis, or α-L-fucosidosis, are characterized by defective degradation of the carbohydrate component of N-linked glycoproteins. In α-mannosidosis, a historically important entity in Angus cattle, a defective enzyme leads to decreased lysosomal α-mannosidase activity in all cells except hepatocytes, which leads to widespread mannose/N-acetylglucosamine oligosaccharide deposition. β-mannosidosis due to β-mannosidase deficiency is reported in Salers cattle and Nubian goats. In both α- and β-mannosidoses, neurons, macrophages and secretory epithelial cells are most severely affected, although storage material is typically lost during tissue processing so vacuoles appear empty on standard H&E slides. In α-L-fucosidosis deficient activity of α-L-fucosidase leads to a similar histological appearance; this condition is autosomal recessive in English springer spaniels.(5)
Mucopolysaccharidoses are distinguished by defective catabolism of glycosaminoglycans, so skeletal and connective tissue abnormalities such as deformities, degenerative joint disease, and thickening of the heart valves or leptomeninges are often observed; neurons can be involved as well. Deficiency in α-L-iduronidase, known as mucopolysaccharidosis type I (MPS I) in humans, is reported in domestic shorthair cats and Plott hounds. Storage primarily occurs in mesoderm-derived cells. Deficiency in N-acetylglucosamine-6-sulfatase leads to the veterinary counterpart of human MPS III, which has been described in Nubian goats. Mesoderm-derived cells are packed with heparan sulfate, while neurons contain gangliosides, which accumulate due to interference with neuraminidase activity. Siamese and domestic shorthair cats occasionally have a deficiency in arylsulfatase-B, a disorder known as MPS VI in humans. Mucopolysaccharidosis VI differs from MPS I and II in that neuronal storage does not occur. Finally, the counterpart to human MPS VII is reported in dogs and cats secondary to a lack of β-glucuronidase. Again, microscopic findings are similar to those described above; however, there is also widespread neurovisceral storage.(5)
Glycogenoses result from defective glycogen catabolism. α-1,4-glucosidase deficiency, an autosomal recessive condition documented in shorthorn and Brahman beef cattle, leads to widespread glycogen storage, both within lysosomes and intracytoplasmically. In contrast, other types of glycogen storage diseases are concentrated primarily within the liver and muscle. Since the excess storage material is composed of glycogen, it is PAS-positive and diastase-sensitive. Neurons are severely affected in this disease.(5)
Acquired lysosomal storage diseases often follow the ingestion of toxic plants, or, less commonly, drug administration. Swainsonine is an indolizidine alkaloid found in several plant species, such as locoweed (Astragalus, Oxytropis sp.). Ingestion of swainsonine by grazing livestock causes inhibition of α-mannosidase, inducing a form of α-mannosidosis. Ingestion by pregnant sheep can also result in abortion and fetal malformation. Ingestion of Trachyandra divaricata or T. laxa causes excessive storage of lipofuscin in central and peripheral neurons of South African/Australian livestock.(5) Aminoglycosides, such as gentamicin, accumulate within lysosomes of renal proximal tubular cells where they inhibit lysosomal phospholipases, producing aggregates of phospholipid-containing myeloid bodies. As lysosomes become progressively distended with myeloid bodies, they rupture, releasing acid hydrolases as well as high concentrations of aminoglycosides into the cytoplasm, further damaging the cell.(4)
Regardless of the underlying genetic or acquired cause, most lysosomal storage diseases in veterinary species are characterized clinically by an early onset of neurologic impairment. Considering the similarity of clinical signs as well as the frequent overlap of the gross and microscopic lesions in many types of lysosomal storage diseases, electron microscopy and especially measurement of lysosomal enzyme activity are often necessary to elucidate a specific etiology.
Table 2: Select inherited lysosomal storage diseases(5)
| Condition | Enzyme Defect | Storage Material | Inheritance/species |
| GM1 gangliosidosis | β-galactosidase | GM1 ganglioside in lysosomes of neurons, glial cells, macrophages | - autosomal recessive - dogs, cats, Friesian cattle; - suffolk sheep - deficiencies in β1-galactosidase AND α-neuraminidase. |
| GM2 gangliosidosis (Tay-Sachs and Sandhoff diseases) | -hexosaminidase (αβ- or ββ-dimer) -activator protein | GM2 ganglioside in lysosomes of neurons, glial cells, macrophages | - autosomal recessive 1. domestic and Korat cats, German shorthaired pointers, golden retrievers: β-subunit deficiency 2. Japanese spaniel dogs, Yorkshire pigs: activator protein deficiency |
| Sphingomyelinosis (Niemann-Pick disease) | sphingomyelinase | sphingomyelin in lysosomes of neurons and macrophages | - autosomal recessive in cat and dog |
| Globoid cell leukodystrophy (glactocerebrosidosis) | galactocerebrosidase | galactocerebrosides in oligodendrocytes/Schwann cells, globoid cell macrophages (NOT in neurons) demyelination, axonal loss | - autosomal recessive - dogs, cats and polled Dorset sheep |
| Glucocerebrosidosis (Gaucher disease) | glucocerebrosidase | glucocerebroside in lysosomes of hepatic/lymph node sinusoidal macrophages, some neurons (NOT in Purkinje cells or the spinal cord) | - Sydney Silky Terriers |
| α-Mannosidosis | α-mannosidase | mannose/N-acetylglucosamine oligosaccharide in lysosomes of neurons, macrophages, secretory epithelial cells | - Angus cattle |
| β-Mannosidosis | β-mannosidase | oligosaccharides in lysosomes of neurons, macrophages, secretory epithelial cells | - Salers cattle and Nubian goats |
| α-L-fucosidosis | α-L-fucosidase | - fucose containing glycoconjugates in lysosomes of neurons - similar appearance to α/β-Mannosidosis | - autosomal recessive
- English springer spaniels |
| MPS I | α-L-iduronidase | mucopolysaccharide storage in mesoderm derived cells | - domestic shorthair cats and Plott hounds |
| MPS III | N-acetylglucosamine-6-sulfatase | heparan sulfate in mesoderm-derived cells; neurons contain gangliosides | - Nubian goats |
| MPS VI | arylsulfatase-B | mucopolysaccharide storage in mesoderm derived cells; neuronal storage does not occur | - Siamese and domestic shorthair cats |
| MPS VII | β-glucuronidase | widespread neurovisceral storage | - dogs and cats |
| Glycogenosis (type II in humans) | α-1,4-glucosidase | widespread glycogen storage within lysosomes and intracytoplasmically; including neurons | - autosomal recessive in shorthorn and Brahman beef cattle |
References:
2. Bundza A, Lowden JA, Charlton KM. Niemann-Pick disease in a poodle dog. Vet Pathol. 1979;16:530-538.
3. Jolly RD, Walkley SU. Lysosomal storage diseases of animals: an essay in comparative pathology. Vet Pathol. 1997;34:527-548.
4. Kaloyanides GJ. Drug-phospholipid interactions: role in aminoglycoside nephrotoxicity. Ren Fail. 1992;14(3):351-357.
5. Maxie MG, Youssef S. Nervous system. In: Maxie MG, ed. Jubb, Kennedy and Palmers Pathology of Domestic Animals. 5th ed. Vol 1. St. Louis, MO: Elsevier; 2007:322-332, 381.
6. Saunders GK, Wenger DA. Sphingomyelinase deficiency (Niemann-Pick disease) in a Hereford calf. Vet Pathol. 2008;45:201-202.
7. Stanbury JB, Wyngaarden JB, Fredrickson DS. The Metabolic Basis of Inherited Disease. 3rd ed. New York, NY: McGraw-Hill; 1972.
8. Wenger DA, Sattler M, Kudoh T, Snyder SP, Kingston RS. Niemann-Pick disease: a genetic model in Siamese cats. Science. 1980;208:1471-1473.