Signalment:
Gross Description:
Histopathologic Description:
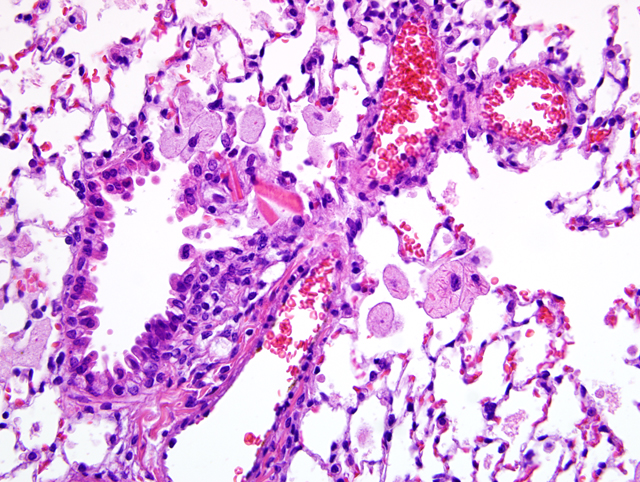
Lung: There is a moderate to marked diffuse intraalveolar infiltrate of macrophages, the cytoplasm of which is moderately to markedly distended with foamy vacuolations (lipid). Many macrophages also contain eosinophilic crystalline material within their cytoplasm, and similar material is present extracellularly within alveolar spaces. Occasional multinucleated giant cells and neutrophils are present, particularly in more severely affected areas. In more severely affected areas, there is an intraalveolar infiltrate of fibrin, and in the most severely affected areas, alveoli are filled with macrophages, crystalline material, and fibrin, with loss of air spaces. Interstitial connective tissue is moderately expanded by edema.Â
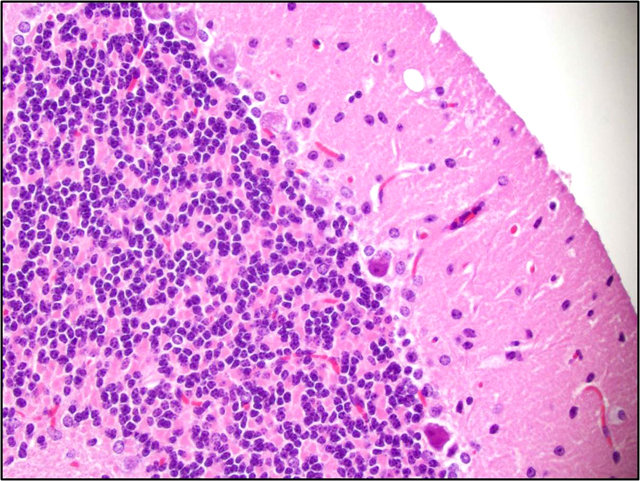
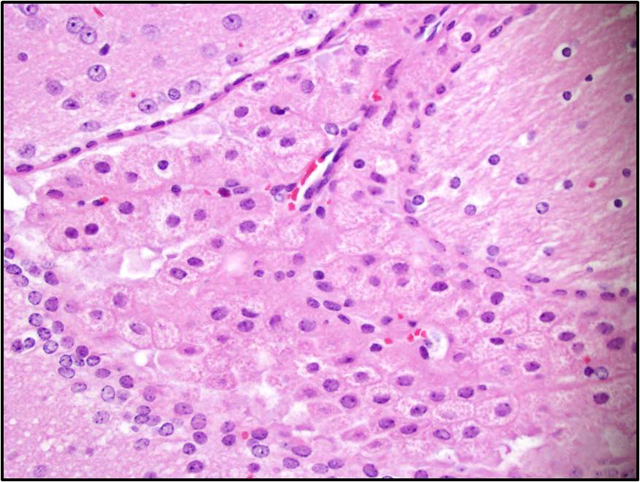
Brain: There is a moderate to marked loss of Purkinje cells that is most severe in the anterior lobes of the cerebellum. Some remaining Purkinje cells are undergoing degeneration and necrosis, with shrinkage, hypereosinophilia, and nuclear pyknosis. Most of the remaining Purkinje cells and many other neurons throughout the brain contain well-delineated, small, lipid-containing vacuoles. There is mild to moderate multifocal gliosis, with an infiltrate of lipid-laden gitter cells. Many choroid plexus cells also contain lipid-laden vacuoles within their cytoplasm.Â
Infiltrates of vacuolated macrophages were also present within the bone marrow, liver, adrenal gland, thymus, thyroid, epididymis, preputial gland, mesenteric lymph node, and myocardium. Vacuolation was also present within ganglion cells adjacent to the heart and neurons within the spinal cord, as well as follicular epithelial cells of the thyroid, epithelial cells of the epididymis, and renal tubular epithelial cells.
Morphologic Diagnosis:
1. Lung: Alveolar histiocytosis, diffuse, moderate to marked, with intracytoplasmic lipid.
2. Lung: Eosinophilic crystalline pneumonia, regionally extensive, moderate, with intraalveolar fibrin.
3. Brain, cerebellum: Purkinje cell necrosis and loss, diffuse, moderate to marked.
4. Brain: Neuronal vacuolation, diffuse, moderate.
5. Brain: Gliosis, multifocal, mild to moderate, with gitter cells containing intracytoplasmic lipid.
Lab Results:
- Hematology:
- Manual Differential:
- Manual Neutrophil %: 46 (3-22.5)
- Band %: 3
- Manual Lymphocyte %: 29 (69-93.5)
- Manual Monocyte %: 4 (0-8)
- Others %: 18
- WBC Morphology (reported by technician): Atypical cells that are classified as others are variable in size ranging from 5 to 20 um. The nucleus of these cells is irregular in shape. Some are angular, others indented, and some are folded. The nuclear chromatin material is clumped in some of the cells and smooth in others.
- Cytoplasm varies from being scant to moderate. In some of the cells the cytoplasm resembles that of a monocyte.
- RBC Morphology: RBC morphology is within normal limits.
- Platelet Morphology: No abnormality noted.
- CBC with automatic differential:
- Red Blood Cells: 9.04 (8.2-10.4)
- Hemoglobin: 14.6 (14.5-16.2)
- Hematocrit: 41.7% (38.5-46.0)
- MCV: 46.1 fL (42-48)
- MCH: 16.1 pg (14.5-16.2)
- MCHC: 35.0 g/dL (31.4-36.8)
- RDW: 19.8% (13.8-17.0)
- Platelets: 445 K/uL (799-1,300)
- Chemistry (liver panel):
- ALP: 83 IU/L (23-181)
- ALT (SGPT): 110 IU/L (16-58)
- AST (SGOT): 1551 IU/L (36-102)
- GGT: 0.0 IU/L (0-2)
- Total Bilirubin: 0.3 mg/dl (0.0-0.3)
- Total Protein: 5.1 g/dl (4.1-6.4)
- Albumin: 3.2 g/dl (2.5-3.9)
- Blood Urea Nitrogen: 27 mg/dl (14-32)
- Creatinine: 0.3 mg/dl (0.1-0.6)
- B/C Ratio: 90.0 (21-127)
Condition:
Contributor Comment:
In humans, an inherited (autosomal recessive) deficiency of ASM activity results in A and B forms of Niemann-Pick disease (NPD).(4,7) Over a dozen mutations cause defects in the ASM enzyme leading to NPD types A and B. Type A NPD is a severe neurodegenerative disorder leading to death by 3 years of age. Individuals with this form of the disease have very little (<5%) ASM activity. In type B NPD, there is little to no neurological involvement, and individuals often survive into adulthood. The decreased severity of clinical signs in these patients is thought to be due to variable amounts (>5%) of ASM activity.(7) A third form of NPD, type C, is also a lipid storage disease, but is due to mutation in unrelated genes (called NPC-1 or NPC-2).Â
In ASMKO mice, the disease is also autosomal recessive, and the phenotype of affected mice is essentially identical to humans with type A NPD.(4) Mice are normal at birth, with ataxia and tremors beginning at approximately 8 weeks of age. There is rapid disease progression, with lethargy, decreased responses to stimuli, and difficulty eating by 12-16 weeks of age. Severe ataxia is also generally present by 16 weeks of age. Death usually occurs by 6-8 months of age. Biochemical analysis of homozygous ASMKO mice reveals no detectable ASM activity in any tissues tested (i.e. liver, lung, spleen, kidney, heart, brain). Lipid analysis of liver and brain showed approximately 15 and 5-fold increases, respectively in sphingomyelin. Total blood cholesterol levels are elevated approximately 80% in these mice, and nearly all of this is associated with the high density lipoprotein (HDL) fraction. Heterozygotes have approximately 50% ASM activity and present with no clinical disease.Â
Reported gross pathology findings in ASMKO mice at the time of naturally-occurring death included a hunched appearance, 50% loss in body and brain weight, and hepatosplenomegaly. Histopathologically, lipid-laden foam cells (so-called NPD cells) were present in most major organs. Electron microscopic evaluation revealed multilamellar cytoplasmic inclusions in all tissues, especially the brain. Additionally, Purkinje cell loss and atrophy of the cerebellum and midbrain were present. The neuronal activity of Purkinje cells far exceeds most other cell types in the CNS, and they form excitatory and inhibitory synapses and relay sensory information from all parts of the body.(7) This high activity level leads to rapid membrane turnover, and this has been hypothesized to account for their increased rate of death in comparison with other neurons in the CNS. This hypothesis is supported by the observation that Purkinje cell loss occurs in the anterior lobules of the cerebellum first, with preservation of the posterior lobular neurons until late in the course of the disease. Posterior lobular Purkinje cells contain high levels of sphingosine kinase, which may serve as an alternate mechanism for sphingolipid breakdown.Â
In addition to neuronal disease, both humans with types A and B NPD and ASMKO mice are reported to develop lung pathology. In human type A NPD, neurodegeneration usually causes death before pulmonary disease becomes advanced. However, individuals with the type B form may show progressive pulmonary dysfunction and respiratory infections that can be fatal.(5) Pulmonary disease is primarily related to alterations of surfactant, which contains increased saturated phosphatidylcholine and sphingomyelin compared to normal individuals. This leads to decreased catabolism by alveolar macrophages (a process that requires lysosomal activity), and subsequent decreased clearance from the lungs. In addition to elevated levels of surfactant, the increased sphingomyelin contributes to abnormal surfactant function. Pulmonary macrophage dysfunction is also thought to play a role in lung disease.(1) Decreased superoxide production by pulmonary macrophages has been documented in ASMKO mice and the addition of ASM in vitro increased superoxide production, further supporting the role of ASM in macrophage function. Additionally, ASMKO mice had elevated levels of macrophage inflammatory proteins (MIP), specifically MIP1_ (a monocyte chemoattractant) and MIP2 (a neutrophil chemoattractant). These chemokines are normally produced by macrophages and bronchial epithelial cells, leading to increased inflammatory cell recruitment. Histopathologically, alveolar macrophages were most affected by the accumulation of intracellular lipids, but ciliated cells of airways, type I pneumocytes, and endothelial cells were also affected. Interestingly, there was no evidence of fibrosis within affected lungs of ASMKO mice, indicating preservation of normal architecture and potential for normal function if ASM activity is restored.
Eosinophilic crystalline pneumonia commonly occurs in strains on a C57BL/6 background.(3) It may occur spontaneously or in concert with other pulmonary lesions, such as pulmonary adenomas, lymphoproliferative disease, allergic pulmonary disease, and parasitic or fungal infection. The crystals in eosinophilic crystalline pneumonia are composed predominately of Ym1 protein, a chitinase-like protein associated with neutrophil granule products and secreted by activated macrophages. The function of Ym1 protein is not fully understood but is believed to be involved in host immune defense, eosinophil recruitment, and cell-cell and cell-matrix interactions consistent with tissue repair. The mechanism of induction of eosinophilic crystalline pneumonia is unknown.Â
JPC Diagnosis:
1. Lung: Alveolar histiocytosis, multifocal, moderate, with intrabronchiolar and intrahistiocytic eosinophilic crystalline material and histiocytic lipid-type cytoplasmic vacuolation.
2. Brain, cerebellum: Purkinje cell degeneration, necrosis, and loss, diffuse, moderate, with gliosis, and neuronal and glial lipid-type cytoplasmic vacuolation.
3. Brain, cerebrum: Neuronal degeneration, necrosis, and loss, diffuse, marked, with gliosis, and neuronal and glial lipid-type cytoplasmic vacuolation.
Conference Comment:
| Macromolecule Class | Lysosomal Storage Disease |
| Sphingolipidoses | GM1 gangliosidosis (generalized gangliosidosis)
GM2 gangliosidosis Glucocerebrosidosis (Gaucher disease) Sphingomyelinosis (NPD Types A, B, and C) Galactosialidosis Galactocerebrosidosis (globoid cell leukodistrophy, Krabbe disease) |
| Glycoproteinoses | -Mannosidosis
-Mannosidosis -L-fucosidosis |
| Mucopolysaccharidoses | Mucopolysaccharidosis types I, III, VI, VII, and others |
| Glycogenoses | Glycogenosis type II (Pompe disease)
Glycogenosis type III (Cori disease) Glycogenosis type IV (Polyglucosan body disease) |
| Mucolipidoses | Inclusion-cell disease (mucolipidosis II) |
| Ceroid lipofuscinoses | Neuronal ceroid-lipofuscinosis (Batten disease)
(see WSC 2008-2009, Conference 24, Case III) |
| Miscellaneous | Lafora disease
Motor neuron disease of English Pointers |
In addition to the C57BL/6 strain, other laboratory mice commonly affected by eosinophilic crystalline pneumonia include 129SvJae and its derivatives, severe combined immunodeficiency (SCID) mice, and knockout mice targeting specific components of the immune system.(3) An example of eosinophilic crystalline pneumonia is available in WSC 2007-2008, Conference 8, Case I.
Focally affecting some sections of lung in this case, alveoli contain small amounts of necrotic cellular debris admixed with low numbers of viable and degenerate neutrophils and macrophages, and the adjacent pleura and alveolar septa are infiltrated by lymphocytes, macrophages, and neutrophils.
References:
2. Gulbins E, Dreschers S, Wilker B, Grassme H: Ceramide, membrane rafts, and infections. J Mol Med 82:357-363, 2004
3. Hoenerhoff MJ, Starost MF, Ward JM: Eosinophilic crystalline pneumonia as a major cause of death in 129S4/SvJae mice. Vet Pathol 43:682-688, 2006
4. Horinouchi K, Erlich S, Perl DP, Ferlinz K, Bisgaier CL, Sandhoff K, Desnick RJ, Stewart CL, Schuchman EH: Acid sphingomyelinase deficient mice: a model of types A and B Niemann-Pick disease. Nature Genetics 10:288-293, 1995
5. Ikegami M, Rajwinder D, Schuchman E: Alveolar lipoproteinosis in an acid sphingomyelinase-deficient mouse model of Niemann-Pick disease. Am J Physiol Lung Cell Mol Physiol 284:L518-L525, 2003
6. Jolly, RD, Walkley SU: Lysosomal storage diseases of animals: an essay in comparative pathology. Vet Pathol 34:527-548, 1997
7. Macauley SL, Sidman RL, Schuchman EH, Taksir T, Stewart GR: Neuropathology of the acid sphingomyelinase knockout mouse model of Niemann-Pick: A disease including structure-function studies associated with cerebellar Purkinje cell degeneration. Experimental Neurology 214:181-192, 2008
8. Maxie MG, Youssef S: Nervous system. In: Jubb, Kennedy, and Palmers Pathology of Domestic Animals, ed. Maxie MG, 5th ed., vol. 1, pp. 322-330. Elsevier Saunders, Philadelphia, PA, 2007
9. Otterbach B, Stoffel W: Acid sphingomyelinase-deficient mice mimic the neurovisceral form of human lysosomal storage disease (Niemann-Pick disease). Cell 81:1053-1061, 1995
10. Saunders GK, Wegner DA: Sphingomyelinase deficiency (Niemann-Pick disease) in a Hereford calf. Vet Pathol 45:201-202, 2008
11. Schuchman EH: The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J Inherit Metab Dis 30:654-663, 2007
12. Zachary JF: Nervous system. In: Pathologic Basis of Veterinary Disease, eds. McGavin MD, Zachary JF, 4th ed., pp. 928-930. Mosby Elsevier, St. Louis, MO, 2007