Joint Pathology Center
Veterinary Pathology Services
Wednesday Slide Conference
2019-2020
Conference 19
19 February, 2020
Dr.
Molly Church, DVM, PhD, DACVP
Assistant Professor
Pathobiology
University of Pennsylvania School of Veterinary Medicine
Philadelphia, PA
CASE II: 18-0311 (JPC 4136399).
Signalment: 4-year-old male castrated mixed breed dog (Canis familiaris)
History: One year prior to euthanasia, the patient began exhibiting bruxism in periods of stress. This worsened to more frequent periods of anxiety and loud bruxism persisting for hours. Five months prior to euthanasia (seven months after onset of clinical signs), the patient became hyperreactive to stimuli around the face and head, showed unclassified ataxia and balance problems, and began to walk into objects. On presentation to the Neurology Service at the university, the patient had vestibular/cerebellar ataxia and an inconsistent menace response bilaterally. Fundic examination was normal. The MRI report stated that the cerebrum had widened cerebral sulci, a severely dilated ventricular system, and small basal nuclei, thalamus, and cerebellum with prominent folia. The patient?s clinical signs continued to progress, and euthanasia was elected due to quality of life concerns.
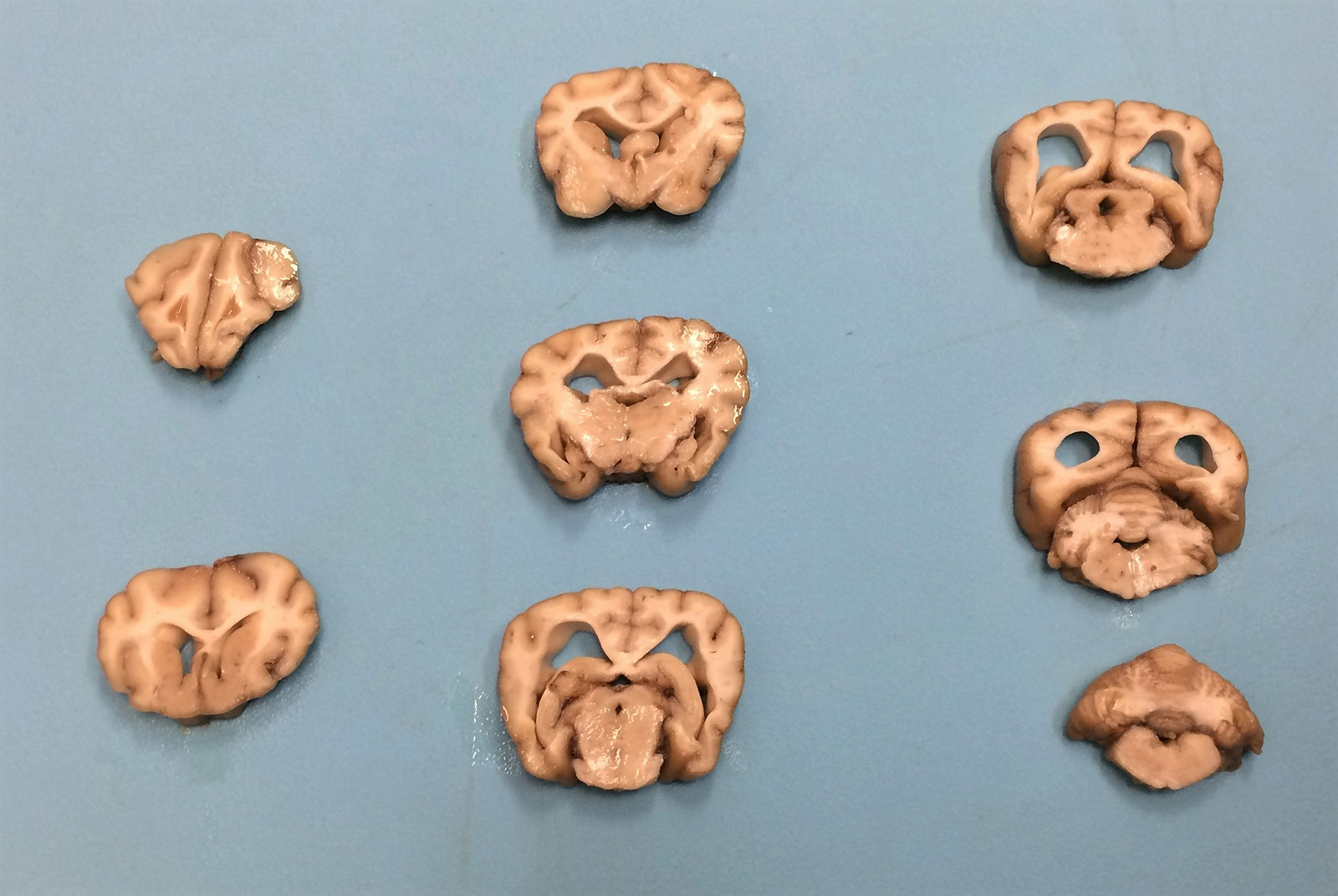
Gross Pathology: The cerebral cortex was severely atrophied with widening of the sulci and secondary dilation of the lateral ventricles (hydrocephalus). The cerebellum appears slightly small. No additional abnormalities were detected on gross examination.
Laboratory results: None.
Microscopic Description:
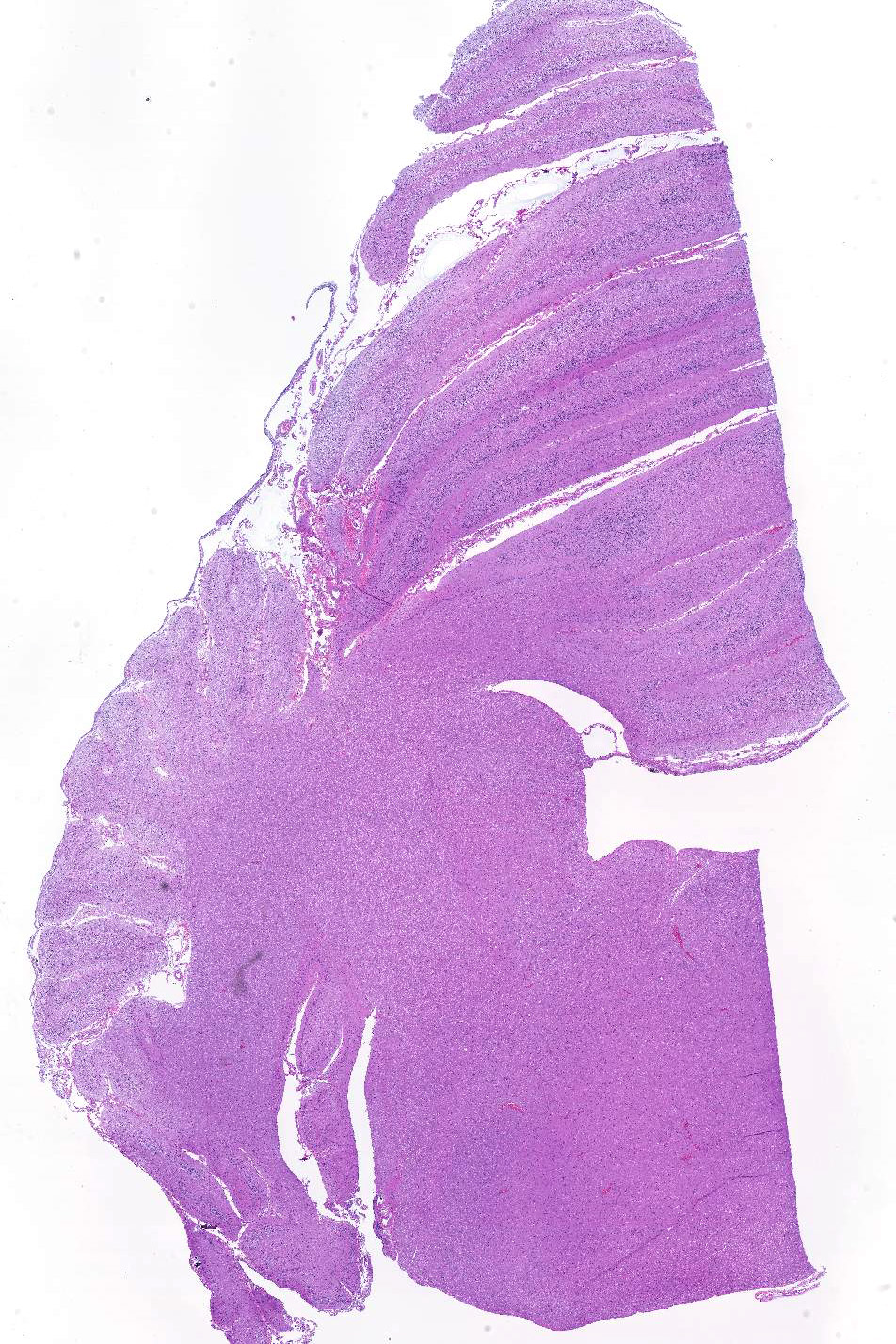
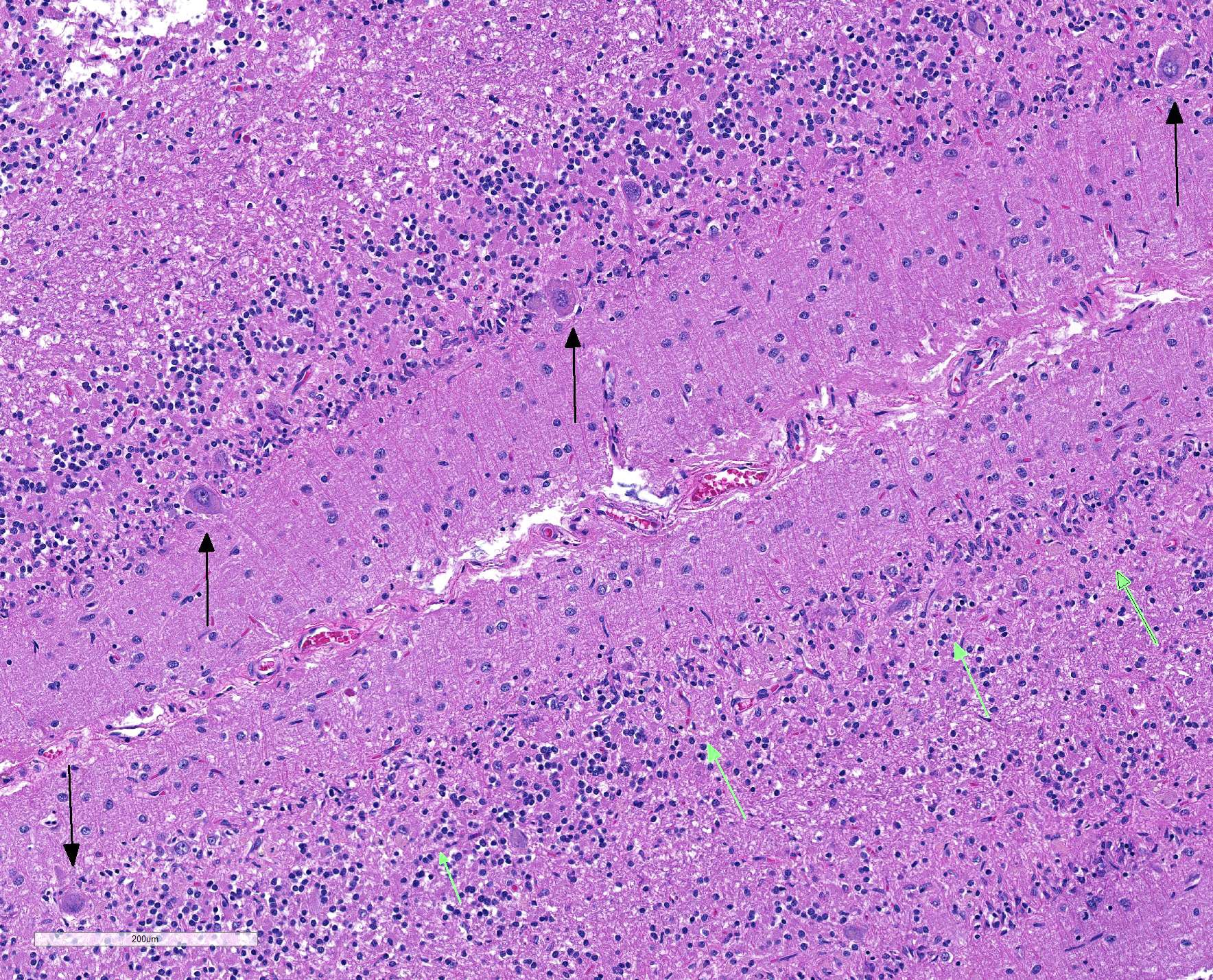
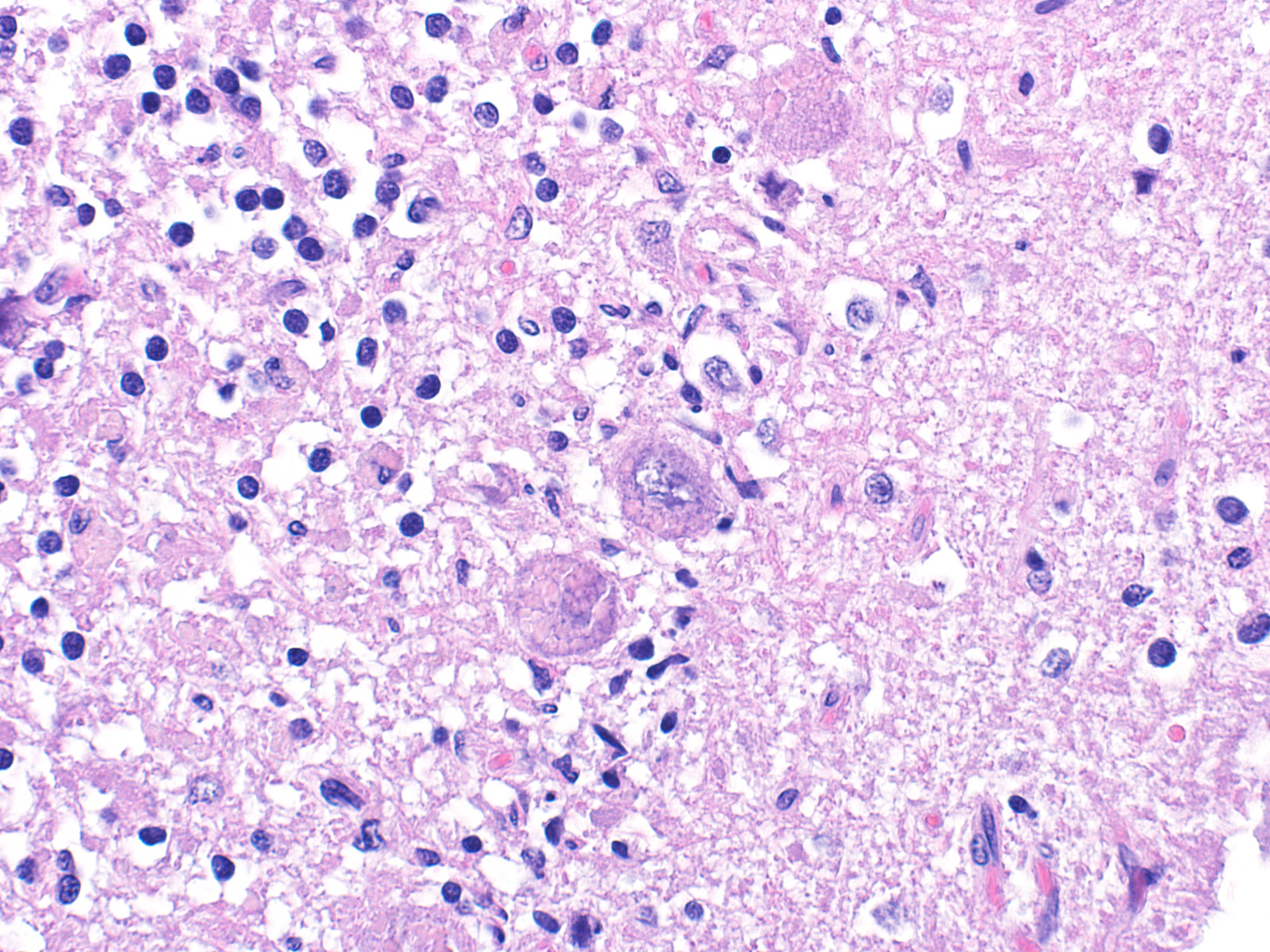
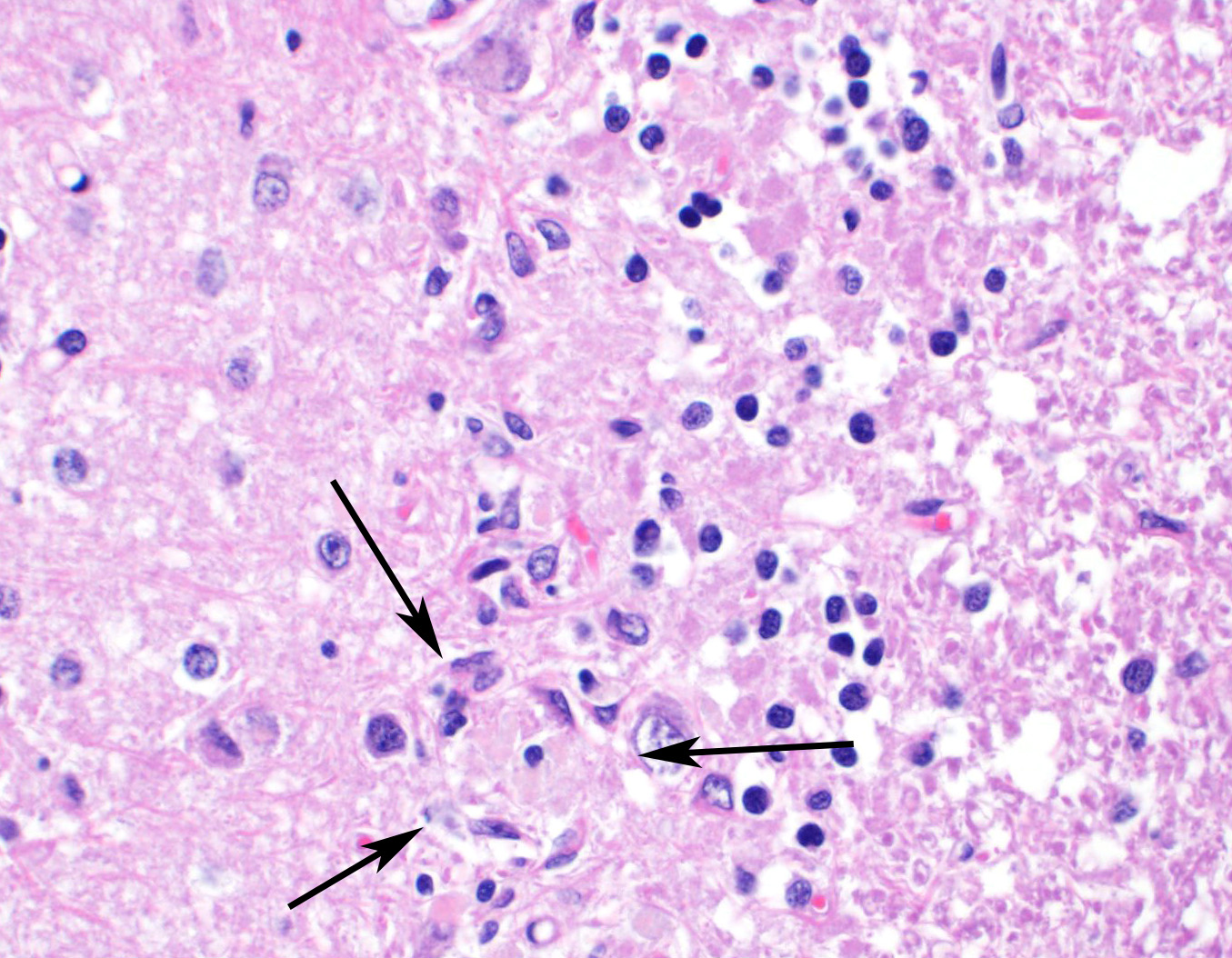
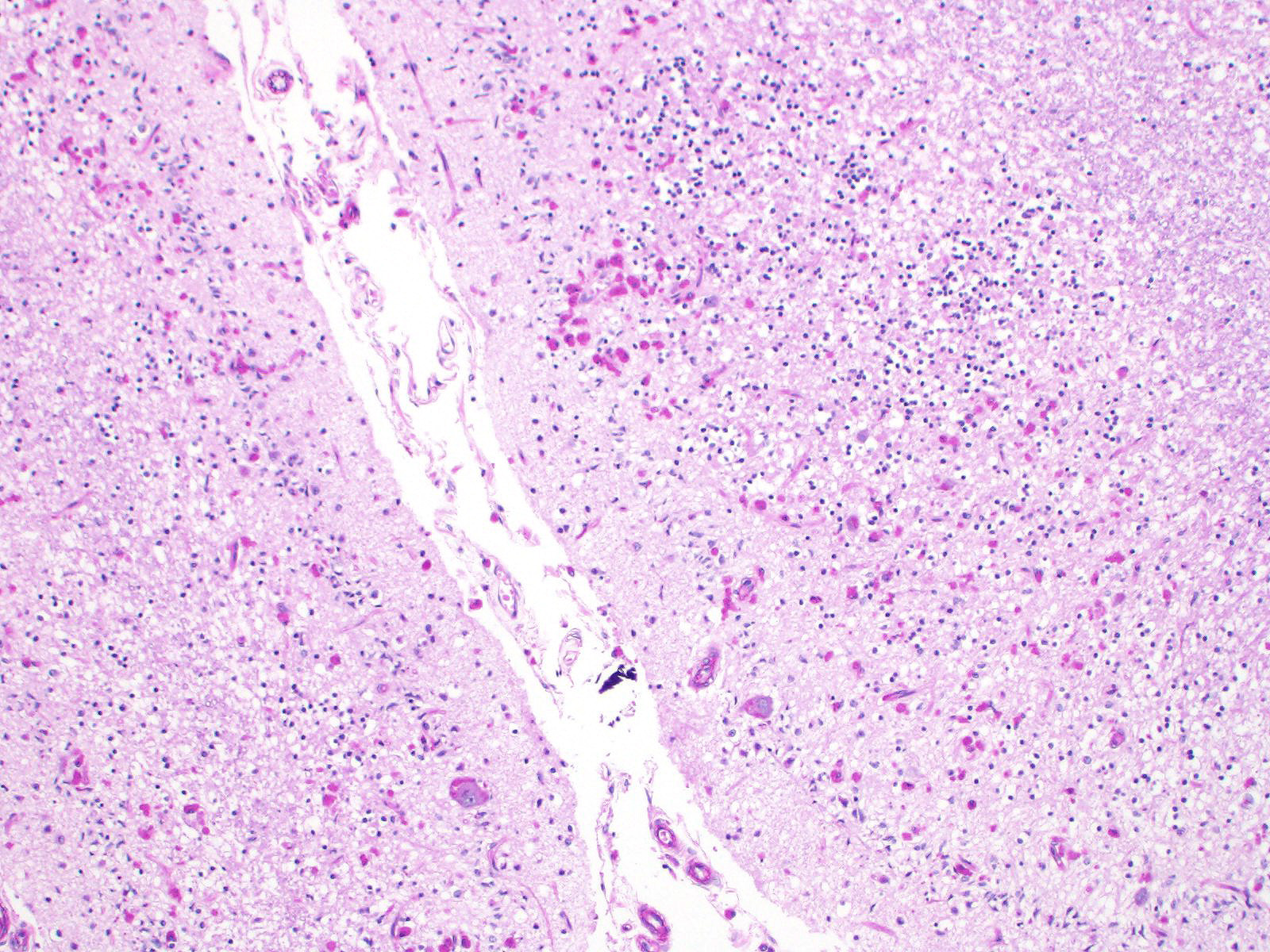
Sections of cerebellum or cerebellum with brainstem are submitted. The cerebellar cortex is mildly diffusely atrophied, with slight thinning and marked pallor of the cerebellar folia. Within the folia, there is marked loss of neurons within the granule cell layer, with fine vacuolation of the remaining parenchyma. Purkinje cells are irregularly spaced, with scattered necrosis and loss. Neurons and glial cells frequently contain abundant pale eosinophilic cytoplasmic storage material that has a globular or granular appearance. The material often peripheralizes the nucleus and variably distends the perikaryon. Affected neurons are degenerate, with cytoplasmic swelling and central chromatolysis. The neuroparenchyma is mildly hypercellular, with increased numbers of glial cells, predominately microglia. Similar changes are detected in neurons throughout the entire central nervous system (cerebral cortex, cerebellum, brainstem nuclei, and spinal cord grey matter) and retina (slides not submitted).
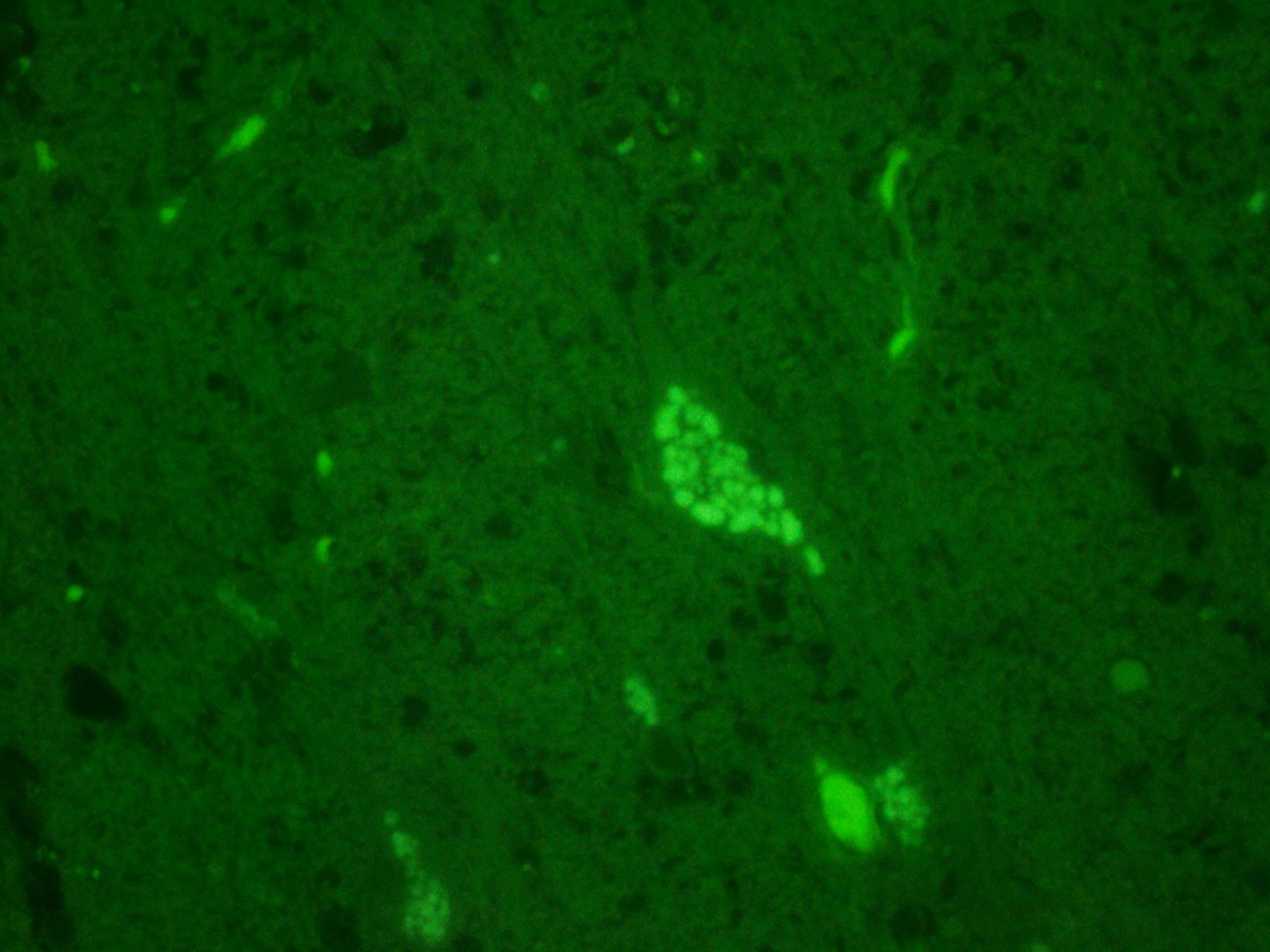
Histochemical stains are applied to multiple sections of central nervous system tissue (see photomicrographs). The cytoplasmic storage material stains magenta with Periodic acid-Schiff (PAS) and is positive with Luxol fast blue.
Contributor Morphologic Diagnosis:
Cerebellum: Severe chronic neuronal degeneration and loss with abundant intracytoplasmic storage material and cortical atrophy
Contributor Comment: Although the recognition of intracellular storage material can be relatively straightforward, definitive identification of the material and subsequent categorization of the storage disease can pose a diagnostic challenge for veterinary pathologists. In this case, an extensive histologic survey of the major organs identified intracytoplasmic pigment only in the central nervous system (CNS) and retina, with the neurons most severely affected. The microscopic appearance, histochemical staining pattern, and cellular distribution of the pigment are most consistent with a type of neuronal ceroid lipofuscinosis (NCL), although definitive diagnosis requires electron microscopy and/or genetic testing.3,6,13
NCLs are neurodegenerative diseases characterized by the accumulation of lipopigment material within cells, always and most severely affecting neurons.3 Like other lysosomal storage diseases, a mutation in a protein (typically an enzyme) critical to the metabolic pathway of digesting a material leads to accumulation of the now indigestible material in residual bodies, gradually leading to cell dysfunction and death.6,20 More than 360 mutations in over a dozen genes have been identified as causes of these diseases in humans8,20,21, dogs1,2,9,10,13, sheep22, pigs4, horses23, goats7, and cattle10. NCL has been described in cats, although a genetic cause has not been identified in this species.5 The vast majority of mutations are within genes coding for lysosomal enzymes, although endoplasmic reticulum and Golgi apparatus proteins have also been implicated.13,15,16,20,21
The accumulated ceroid-lipofuscin lipopigment in NCL is similar to both ceroid and lipofuscin but is not truly a variant of either. Ceroid is a pigment that accumulates within cells due to a pathological process, such as a nutritional deficiency. Lipofuscin is also a pigment that accumulates within post-mitotic cells with age as a ?wear-and-tear? material. The NCL lipopigment is composed primarily of protein with lesser lipid components.13,15 The specific protein component is determined by mutation, but is typically derived from subunit C of mitochondrial ATP synthase13, or less commonly from a sphingolipid activator protein.18,20
Ceroid-lipofuscin is yellow-gold to lightly eosinophilic on H&E staining. In cases of NCL, there are globular or botryoid accumulations within the axon hillock that displace the nucleus and Nissl substance. The material stains positively with PAS and Luxol fast blue stains and is variably acid-fast positive.3 Ultrastructurally, the lipopigment has characteristic ?curvilinear? or ?fingerprint bodies? approximately 15 nm in diameter.3,6,13,20 The material will autofluoresce under ultraviolet light, particularly in unstained paraffin-embedded tissue sections13,15
Neurons throughout the CNS are always affected, although the material may also accumulate in neurons of the peripheral nervous system neurons (e.g. in the intestinal plexi) as well as other cells in the CNS, including astrocytes, oligodendrocytes, and microglia. Later in disease, cells in tissues outside of the nervous system may also accumulate storage material, including hepatic Kupffer cells and epithelial cells in the kidneys, pancreas, lungs, reproductive organs, skin, endocrine organs, salivary glands, etc.13,16,20
Affected individuals show progressive cognitive decline, visual and motor deficits, and seizures.13,14 Age of onset can vary greatly, with most affected animals showing signs early in life, however there are late forms as well.6,15 The disease is invariably fatal, with death occurring within months or up to a few years after clinical signs first present.3,20
Contributing Institution:
University of Pennsylvania
School of Veterinary Medicine
Department of Pathobiology
http://www.vet.upenn.edu/research/academic-departments/
JPC Diagnosis: Cerebellum: Neuronal degeneration, necrosis, and loss, diffuse, severe, with marked neuronal intracellular granular pigment accumulation, gliosis, and neuronophagia.
JPC
Comment: The
contributor has provided an excellent review of neuronal-ceroid lipofuscinosis
in animals.
The disease was first described in humans by Dr. Otto Christian Stengel in
Germany as a juvenile onset disorder resulting in blindness and progressive
dementia. In 1902, English neurologist and pediatrician Frederick Eusace
Batten described a similar disorder in two members of the same family, but was
also the first to describe the neuropathology of cerebral and ocular macular
degeneration, and it is from this investigator that the disease was called Batten?s
disease for many years (with that term now being restricted to certainly
particular forms of the disease).18
Today, at least 14 affected genes have been implicated in NCL (neuronal
ceroid-lipofuscinosis), which result in various manifestations that may appear
in infants, toddlers, juveniles, and adult onset form. Eight of these 14 genes
have been identified in canine CNL, as noted in Table 1, below.12 A
number of schemes are used in the classification of NCLs in humans, to include
the historical schema, largely based on onset, a classification scheme based on
abnormal genes and accumulated proteins, and one based on typical
ultrastructural findings and abnormal enzymatic activities. A review of these
classifications and a very good overall review of the disease in general in
humans is available by Nita et al. below.18
Although there are a wide diversity of mutated genes, affected proteins, and manifestations, the NCLs are traditionally grouped together due to the common presence of autofluorescent pigment accumulation within neurons and other cells. Like many lysosomal storage diseases, many of the identified abnormal gene products in the variants of NCL accumulate in lysosomes, as do ceroid lipoportain pigments. While early attempts at classification assumed that the appearance of inclusions were specific for each variant, more recent investigation has shown that they are not specific for each disease, may vary with tissue examined, and the same NCL may include more than one pattern of inclusion.18
References:
1. Awano T, Katz ML, O'Brien DP, Sohar I, et al. A frame shift mutation in canine TPP1 (the ortholog of human CLN2) in a juvenile Dachshund with neuronal ceroid lipofuscinosis. Molec Genet Metab. 2006; 89: 254-260.
2. Awano T, Katz ML, O'Brien DP, Taylor JF, et al. A mutation in the cathepsin D gene (CTSD) in American bulldogs with neuronal ceroid lipofuscinosis. Molec Genet Metab. 2006; 87: 341-348.
3. Cantile C, Youssef, S. Nervous system. In: Maxie MG, ed. Jubb, Kennedy, and Palmer?s Pathology of Domestic Animals. Vol 1. 6th ed. St. Louis, MO: Saunders Elsevier; 2016: 290-292.
4. Cesta MF, Mozzachio K, Little PB, Olby NJ, Sills RC, Brown TT. Neuronal ceroid lipofuscinosis in a Vietnamese pot-bellied pig (Sus scrofa). Vet Pathol. 2006;43(4):556-560.
5. Chalkley MD, Armien AG, Gilliam DH, Johnson GS, Zeng R, Wünschmann A, Kovi RC, Katz ML. Characterization of neuronal ceroid-lipofuscinosis in 3 cats. Vet Pathol. 2014;51(4):796-804
6. Ferreira CR, Gahl WA. Lysosomal Storage Diseases. Translational Science of Rare Diseases. 2017; 2: 1?71.
7. Fiske RA, Storts RW. Neuronal ceroid-lipofuscinosis in Nubian goats. Vet Pathol. 1988; 25: 171-173.
8. Gao HL, Boustany RMN, Espinola JA, Cotman SL, et al. Mutations in a novel CLN6-encoded transmembrane protein cause variant neuronal ceroid lipofuscinosis in man and mouse. Am J Hum Genet. 2002; 70: 324-335.
9. Goebel HH, Bilzer T, Dahme E, Malkusch F. Morphological studies in canine (Dalmatian) neuronal ceroid-lipofuscinosis. Am J Med Genet Suppl. 1988; 5: 127-139.
10. Guo J, Johnson GS, Brown HA, Provencher ML. A CLN8 nonsense mutation in the whole genome sequence of a mixed breed dog with neuronal ceroid lipofuscinosis and Australian shepherd ancestry. Molec Genet Metab. 2014; 112: 302-309.
11. Guo J, Johnson GS, Cook J, Harris OK, Mhlanga-Mutangadura T, Schabel RD, Jensen CA, Katz ML. Neuronal ceroid lipofuscinosis in a German Shorthaired Pointer associated with a previously reported CLN8 nonsense variant. Mol Gen Metabol Rep 2019; 21. Htps://doi.org/10.1016/j.ymgm4.2019.100521.
12. Harper PAW, Walker KH, Healy PJ, Hartley WJ, et al. Neurovisceral ceroid-lipofuscinosis in blind Devon cattle. Acta Neuropathologica. 1988; 75: 632-636.
13. Jolly RD, Palmer DN, Studdert VP, Sutton RH, et al. Canine ceroid-lipofuscinoses: A review and classification. Journal of Small Animal Practice. 1994; 35: 299-306.
14. Katz ML, Narstrom K, Johnson GS, O?Brien DP. Assessment of retinal function and characterization of lysosomal storage body accumulation in the retinas and brains of Tibetan terriers with ceroid-lipofuscinosis. Am Jour Vet Research. 2005; 66:67-76.
15. Katz ML, Rustad E, Robinson GO, Whiting REH, Student JT, Coates JR, Narfstrom K. Canine neuronal ceroid lipofuscinoses: Promising models for preclinical testing of therapeutic interventions. Neurobiology of Disease. 2017; 108: 277-287
16. Melville SA, Wilson CL, Chiang CS, Studdert VP. A mutation in canine CLN5 causes neuronal ceroid lipofuscinosis in border collie dogs. Genomics. 2005; 86: 287-294.
17. Nakamoto Y, Yamato O, Uchida K, Nibe K. Neuronal Ceroid-Lipofuscinosis in longhaired chihuahuas: Clinical, pathologic, and MRI findings. J Am Anim Hosp Assoc. 2011; 47: E64-E70.
18. Nita DA, Mole SE, Minassian BA. Neuronal ceroid lipofuscinoses. Epileptic Diosrd 2016; 18(Supple 2):S73-S88.
19. Palmer DN, Tyynela J, vanMil HC, Westlake VJ, Jolly RD. Accumulation of sphingolipid activator proteins (SAPs) A and D in granular osmiophilic deposits in miniature schnauzer dogs with ceroid-lipofuscinosis. J Inherit Metab Dis. 1997; 20: 74-84.
20. Pastores GM, Maegawa GHB. Neuropathic Lysosomal Storage Disorders. Neurol Clin. 2013 November; 31(4): 1051?1071.
21. Sun, A. Lysosomal storage disease overview. Ann Transl Med 2018; 6(24):476.
22. Tammen I, Houweling PJ, Frugier T, Mitchell NL, et al. A missense mutation (c. 184C > T) in ovine CLN6 causes neuronal ceroid lipofuscinosis in Merino sheep whereas affected South Hampshire sheep have reduced levels of CLN6 mRNA. Biochim Biophys Acta. 2006;1762: 898-905.
23. Url A, Bauder B, Thalhammer J, Nowotny N, et al. Equine neuronal ceroid lipofuscinosis. Acta Neuropathologica. 2001; 101: 410-414.