Joint Pathology Center
Veterinary Pathology Services
Wednesday Slide Conference
2011-2012
Conference 25
16 May 2012
CASE II: 11-4158 (JPC 4002911).
Signalment: 6-month-old male intact Shih Tsu mix dog (Canis familiaris).
History: The puppy was presented to the veterinary teaching hospital 15-18 hours after ingesting up to 8 Midol tablets (500mg acetaminophen, 60mg caffeine, 15mg pyrilamine maleate per tablet). No decontamination had been attempted. The patient was treated with N-acetylcysteine and Denamarin, plus Norm-R fluids at ½ maintenance dose. The puppy exhibited signs of hepatic encephalopathy and was euthanized due to poor prognosis.
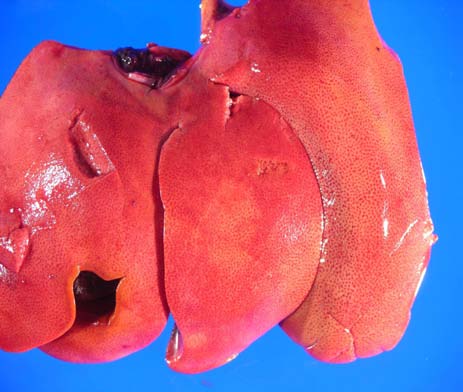
Gross Pathology: There was generalized mild icterus of subcutis and mucous membranes. The liver was diffusely pale tan to yellow with a distinct reticular pattern. At the time of necropsy, the urine was transparent yellow.
Laboratory Results: Presentation:
ALT = 261
AP = 239.
One day later:
ALT = 17, 090
AP = 578
Total bilirubin = 4.5
Urine was coffee colored.
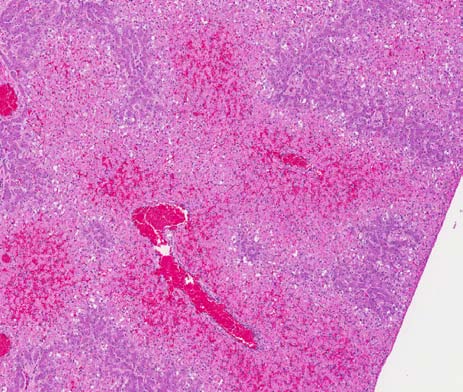
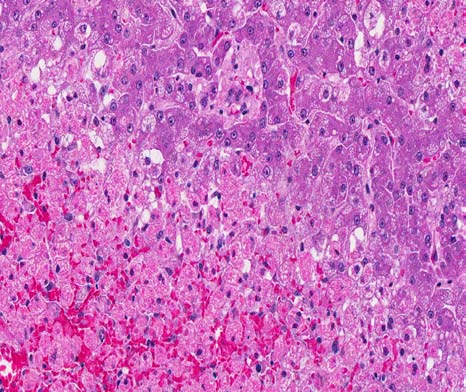
Microscopic Description: Liver: Approximately 50% of the liver parenchyma was involved in acute degenerative to necrotizing changes. There was extensive coagulative necrosis of hepatocytes adjacent to all central veins and extending to the midzonal regions. Necrotic hepatocytes were rounded up and contained hypereosinophilic, fragmented cytoplasm. Scattered cells lacked nuclei, while others contained pyknotic to karyorrhectic nuclei. Sinusoids adjacent to central veins were filled with red blood cells (congestion).
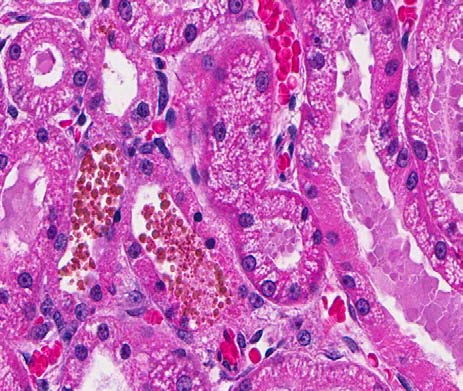
Kidney: Widely scattered tubules within the cortex and medulla contained dark orange-brown globules (presumptive hemoglobin). There were similar pigment globules within the cytoplasm of widely scattered epithelial cells lining these tubules. Proximal convoluted tubular epithelium had scattered degenerate cells with hypereosinophilic cytoplasm and pyknotic nuclei. Regenerative epithelial cells were rare. The urinary spaces of several glomeruli were moderately dilated and filled with weakly eosinophilic protein fluid residue. There was similar protein fluid within numerous tubules within the cortex.
Within the stomach (not shown) were numerous lymphocytic nodules in the lamina propria, associated with large, silver positive, spiral bacteria (presumptive Helicobacter sp.) in the superficial mucus and glands.
Microscopic Diagnosis: 1. Diffuse, acute, severe centrilobular hepatocellular necrosis.
2. Diffuse, mild to moderate hemoglobinuric nephrosis (presumptive).
3. Diffuse, moderate, chronic gastritis (tissue not submitted).
Contributor Comment: The lesions in this puppy were compatible with acute acetaminophen intoxication. The toxic dose of acetaminophen in dogs ranges from 200-600mg/kg6; this 4kg puppy ingested up to 4grams in 8 Midol tablets. The recommended therapeutic dose in dogs is 15mg/kg every 8 hours. By contrast, cats are much more susceptible to toxicity and toxicosis has been seen with doses as low as 10mg/kg.
Acetaminophen (N-acetyl-p-aminophenol) is a widely used over-the-counter analgesic and antipyretic drug considered very safe in humans at therapeutic doses and having fewer gastrointestinal side effects than aspirin or ibuprofen.4 Yet poisoning by acetaminophen accounts for approximately half of all cases of acute liver failure in people in the US and Britain. Poisoning in dogs and cats is usually associated with administration by well-intentioned by uninformed owners, or by accidental ingestion as in this case.6
The liver is a common target of toxicosis, especially from ingested toxins, due to several factors.7 The liver is exposed to high concentrations of ingested compound via the portal circulation; exogenous and endogenous compounds concentrate in the liver by binding to transport proteins and enzymatic sites; and as the primary site of biotransformation of ingested compounds it is exposed to high concentrations of metabolites. Hepatic biotransformation of xenobiotics occurs in 2 phases: phase I involves metabolism generally by mixed function oxidases, and phase II involves detoxification of metabolites by conjugation with water, sulfate, glucuronate, glutathione and others. Toxicity occurs when metabolism into toxic metabolites overwhelms conjugation systems. As such the centrilobular hepatocytes (zone 3), with high concentrations of mixed function oxidases, are most susceptible to toxicity from compounds that are metabolized to toxic intermediates requiring conjugation. Acetaminophen is a classic example of such a compound.
Mechanisms of acetaminophen induced liver injury have been recently reviewed.4 Although still under intense investigation, hepatotoxicity occurs in 5 primary steps: 1) metabolism into reactive metabolite by cytochrome P450 (CYP); 2) consumption of glutathione leading to excess reactive oxygen and nitrogen species; 3) increase oxidative stress resulting in alterations in calcium homeostasis and mitochondrial permeability; 4) loss of mitochondrial ability to synthesize ATP and 5) loss of ATP leading to necrosis.
At non-toxic doses, acetaminophen is metabolized by direct conjugation to glutathione and the non-toxic conjugates are excreted in urine and bile.6 At toxic doses, glutathione is overwhelmed and metabolism is shunted to the CYP system, resulting in the toxic metabolite N-acetyl-p-benzoquinone imine (NAPQI). NAPQI requires glutathione for detoxification; with glutathione already severely depleted, NAPQI binds covalently to cellular proteins and enzymes, disrupting membranes and leading to oxidative stress. Depletion of glutathione also leads to increased reactive oxygen and nitrogen species. N-acetylcysteine, developed for treatment of acetaminophen toxicity, increases detoxification of NAPQI by direct conjugation and through increased glutathione synthesis. This compound, and S-adenosylmethionine (an intermediary in production of cell membranes and glutathione), have been used effectively in dogs and cats with acetaminophen toxicity.6 Although uncoupling of mitochondrial permeability transition with subsequent loss of ATP is considered the primary mechanism of acetaminophen induced hepatotoxicity, other mechanisms that have been explored are altered hepatic blood flow due to necrosis of sinusoidal endothelium, and the role of inflammation, chemokines and cytokines both in development of toxicity and in hepatic repair.4
Methemoglobin formation is the primary mechanism of toxicity in cats; hepatotoxicity occurs only at high doses. Cats are more susceptible to acetaminophen toxicity than dogs because of much lower levels of glucuronyltransferase.6 In addition, feline red blood cells have more sulfhydryl groups than other species, making them highly susceptible to oxidative injury. In contrast, dogs develop methemoglobinemia only at higher toxic doses. Histologic lesions of hemoglobinuric nephrosis in this case, although mild, support the likelihood that this puppy ingested high enough doses of acetaminophen to produce metahemoglobinemia.
The other toxic principle in Midol tablets is caffeine, a methylxanthine in the same class of compounds with theobromine and theophylline.2 Toxic doses for dogs range from 110-200mg/kg; the 4kg puppy ingested up to 480mg, making direct toxicity from caffeine a possible complication. Caffeine is also metabolized by the liver; hepatic damage from acetaminophen could have compromised caffeine metabolism and enhanced its toxicity in this case. Signs of caffeine toxicity are vomiting, tremors and seizures. There are no gross or histologic lesions related to toxicity, although it is possible that the terminal CNS signs in this dog may have been due to a combination of liver failure and caffeine toxicity.
JPC
Diagnosis:
1. Liver: Necrosis, centrilobular, diffuse.
2. Kidney, proximal tubules:
Degeneration and necrosis, multifocal, mild, with hemoglobin casts.
Conference Comment: Heinz bodies, which are foci of denatured globin apparent as a membranous inclusion in the erythrocyte, are expected to be present in a peripheral blood smear in this case due to the oxidative damage to erythrocytes from acetaminophen toxicity, and are an indicator of intravascular hemolysis. Heinz body formation results from oxidative damage that causes disulfide links between glutathione and globin chains, resulting in aggregation and precipitation of globin in the cell. Also, the predicted mean cell hemoglobin concentration (MCHC) would be increased due to artifact from measured Heinz bodies which would artificially increase the MCHC. Luna hemoglobin stain can be used to confirm the hemoglobin casts within the renal tubules.3
The serum hepatic chemistry changes in this case are typical of acetaminophen toxicosis, with normal hepatic enzymes on the first day and massive increases by the third day, which demonstrates the clinical importance of not ruling out hepatic necrosis even when hepatic enzymes are normal when first measured. The reason the alanine transaminase (ALT) is markedly increased and the alkaline phosphatase (AP) is relatively normal the day after presentation is ALT, in addition to aspartate aminotransferase (AST), is a leakage enzyme and increases with hepatic degeneration and necrosis, while AP is an inducible enzyme mainly useful in detecting cholestasis and originates from the biliary tree.1
Conference participants discussed how hemoglobinuric nephrosis is a misnomer, as hemoglobin is not directly nephrotoxic. Hemoglobin passes into the glomerular filtrate after haptoglobin saturation, resulting in formation of granular casts. The combination of ischemia and hypoxia due to anemia and hypotension, as well as tubular obstruction, and interstitial edema contribute to the tubular necrosis. Conference participants discussed the common causes of coffee-colored urine in dogs, and cystitis with concurrent hematuria is the most likely source, with hemoglobinuria being next, and myoglobinuria being very rare.5 Conference participants interpreted the eosinophilic fluid within Bowman’s space as either autolysis or reflux from tubular necrosis.
Contributor: Washington State University
Department of Veterinary Microbiology and Pathology
Pullman, WA 99164-7040
www.vetmed.wsu.edu
References:
1. Bain PJ. Liver. In: Latimer KS, ed. Duncan and Prasseâs Veterinary Laboratory Medicine: Clinical Pathology. 5th ed. Ames, IA: Wiley-Blackwell; 2011:213-23.
2. Carson TL. Methylxanthines. In: Peterson ME, Talcott PA, eds. Small Animal Toxicology, 2nd ed. St Louis, MO: Elsiever; 2006: 845-852.
3. Fry MM, McGavin MD. Bone marrow, blood cells, and the lymphatic system. In: McGavin MD, Zachary JF, eds. Pathologic Basis of Veterinary Disease. 5th ed. St. Louis, MO: Mosby; 2011:705-11.
4. Hinson JA, Roberts DW, James LP. Mechanisms of acetaminophen-induced liver necrosis. In: Utrecht J, ed. Adverse Drug Reactions: Handbook of Experimental Pharmacology. Berlin, Germany: Springer-Verlag; 2010:368-405.
5. Maxie MG, Newman SJ. Urinary system. In: Maxie MG, ed. Jubb, Kennedy, and Palmerâs Pathology of Domestic Animals. 5th ed. Vol. 2. Philadelphia, PA: Saunders Elsevier; 2007:432-6.
6. Sellon RK. Acetaminophen. In: Peterson ME, Talcott PA, eds. Small Animal Toxicology. 2nd ed.St. Louis, MO: Elsiever; 2006: 550-557.
7. Stalker MJ, Hayes MA. Liver and biliary system. In: Maxie MG, ed. Jubb, Kennedy and Palmerâs Pathology of Domestic Animals. 5th ed. Edinburgh, Scotland: Elsevier; 2007:364-367.