Signalment:
Gross Description:
Histopathologic Description:
Morphologic Diagnosis:
Condition:
Contributor Comment:
Histopathologically, LD is characterized by the presence of pathognomonic inclusions named Lafora bodies (LB) especially in the brain, spinal cord and other tissues such as skin, liver, cardiac and skeletal muscle.(2) In the central nervous system LB are localized mostly in the perikarya. These inclusions can become very large, leading to cell death.(1)
LB are carbohydrate storage products that are composed of polyglucosans, which are abnormally formed glycogen molecules that resemble starch. This polyglucosans are poorly branched which makes them insoluble leading to the formation of the characteristic intracellular inclusions.(14)
In most cases this disorder is caused by mutations in one of two different genes, EPM2A and EPM2B (also known as NHLRC1) which encode the proteins laforin and malin, respectively.(3,11) Laforin is a glycogen phosphatase which plays an important role in glycogen metabolism and is involved in dephosphorylation of complex carbohydrates such as amylopectin and glycogen and thus preventing the soluble glycogen molecules from becoming insoluble polyglucosans.(9) Malin on the other hand is an E3 ubiquitin ligase that is believed to target laforin catalyzing its polyubiquitination and thus its degradation. However, the specific role of malin in this process is yet to be determined.(9)
In the dog LD has been reported in the Basset Hound, Miniature Wirehaired Dachshund, Poodle and Beagle also.(8,18) In these animals progressive myoclonus is associated with the presence of intraneuronal storage of complex polyglucosans forming the so-called LB.(8)
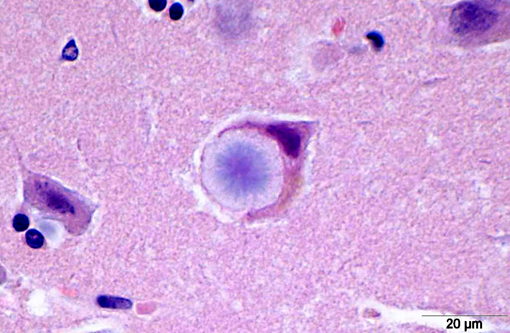
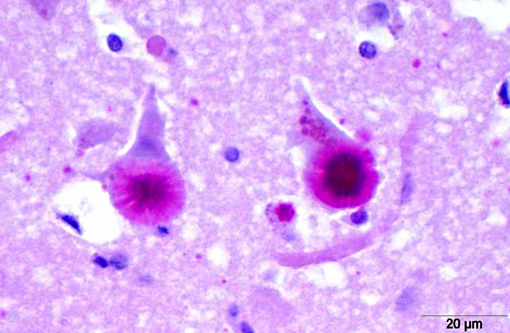
LB in the dog appear as spherical basophilic to amphophilic, mostly intracytoplasmatic inclusions with peripheral radiating filaments; their size may vary from 5 to 20 μm and can be localized in the dendrites, axons or perikarya (unlike in humans).(2) LB may be found anywhere along the neuroaxis and retinal ganglion cells but in dogs with neuronal disease they are most common in the Purkinje cells and neurons of the caudate, thalamic and periventricular nuclei.(18) LB are PAS positive, diastase resistant, stain positively with Bests carmine, methenamine silver, and Weils.(7) In electron microscopy, they are not membrane bound and are composed of fibrillary and electron-dense, sometimes granular components in varying proportions. They are often associated with rough endoplasmic reticulum and Golgi. LB present as accumulations of insoluble complex glycoprotein polymers that need to be distinguished from other types of polyglucosan bodies such as Lafora-like bodies, corpora amylacea, and amylopectin bodies.(14) The distinction between LB and other inclusions is performed based on clinical features and tissue distribution of the inclusions.(15)
Corpora amylacea are inclusions that are formed in normal aging cells and are chemically and histochemically indistinguishable from LB. Corpora amylacea can be found in axons and astrocytic processes but not in the perikarya, unlike LB.(15)
Amylopectin bodies are formed in patients with glycogen storage disease type IV or glycogen branching enzyme deficiency.(19,20) They result from accumulation of abnormal polysaccharides and are mostly indistinguishable from LB in terms of morphology and distribution and thus the clinical presentation of the disease should be used in their distinction.(19,20)
Lafora-like bodies are also very similar in their morphology and distribution but are observed in normal aged animals without neurologic symptoms.(19)
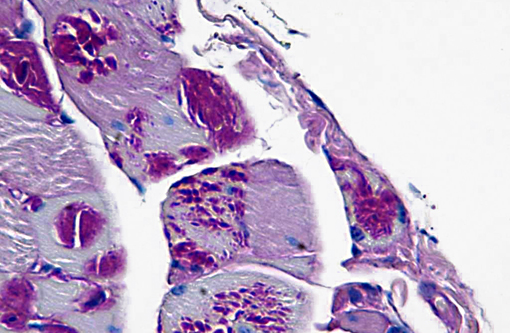
In cases of LD, typically LB are also found in skeletal muscle samples where they lie between myofibrils or beneath the sarcolemma and therefore muscle biopsies can be used for the diagnosis of the disease.(15) In the case presented here, a biopsy of the cranial tibial muscle was submitted for histopathological analysis and accumulations of PAS positive, diastase resistant, brightly staining material forming focal intraparenchymal subsarcolemmal accumulations consistent with LB were found in the skeletal muscle fibers.
The canine counterpart of LD seems to be associated with a mutation in the EPM2B gene, more precisely a dodecamer expansion in the gene.(10) This mutation was observed in 5% of Miniature Wirehaired Dachshunds in England.(10)
The presence of LB associated with neurologic disease has also been documented in cats and cows.(5,16) A recent report described a Lafora-�s-like disease in a fennec fox (Vulpes zerda)(7) and another report demonstrated LB in the brain of an aged captive raccoon (Procyon lotor).(6)
JPC Diagnosis:
Conference Comment:
References:
2. Andrade DM, Turnbull J, Minassian BA. Lafora disease, seizures and sugars. Acta Myol. 2007;26:83-86.
3. Chan EM, Young EJ, Ianzano L, et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat Genet. 2003;35:125-127.
4. Delgado-Escueta AV, Ganesh S, Yamakawa K. Advances in the genetics of progressive myoclonus epilepsy. Am J Med Genet. 2001;106:129-138.
5. Hall DG, Steffens WL, Lassiter L. Lafora bodies associated with neurologic signs in a cat. Veterinary Pathology. 1998;35:218-220.
6. Hamir AN. Spontaneous lesions in aged captive raccoons (Procyon lotor). J Am Assoc Lab Anim Sci. 2011;50:322-325.
7. Honnold SP, Schulman FY, Bauman K, et al. Lafora's-Like Disease in a Fennec Fox (Vulpes Zerda). J Zoo Wildlife Med. 2010;41:530-534.
8. Jubb KVF, Kennedy PC, Palmer NC. Pathology of Domestic Animals. 5th ed. Philadelphia, PA: Saunders Elsevier; 2007;898.
9. Knecht E, Aguado C, Sarkar S, et al. Impaired autophagy in Lafora disease. Autophagy. 2010;6:991-993.
10. Lohi H, Young EJ, Fitzmaurice SN, et al. Expanded repeat in canine epilepsy. Science. 2005;307:81-81.
11. Minassian BA, Ianzano L, Delgado-Escueta AV, et al. Identification of new and common mutations in the EPM2A gene in Lafora disease. Neurology. 2000;54:488-490.
12. Minassian BA. Lafora's disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol. 2001;25:21-29.
13. Sainsbury R. DNA scrrening for Laforas disease in miniature wire-haired dachshunds. Vet Rec. 2011;169:292.Â
14. Sakai M, Austin J, Witmer F, et al. Studies in myoclonus epilepsy (Lafora body form). II. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology. 1970;20:160-176.
15. Schoeman T, Williams J, van Wilpe E. Polyglucosan storage disease in a dog resembling Lafora's disease. J Vet Intern Med. 2002;16:201-207.
16. Simmons MM. Lafora Disease in the Cow. Journal of Comparative Pathology 1994;110:389-401.
17. Singh S, Ganesh S. Lafora progressive myoclonus epilepsy: a meta-analysis of reported mutations in the first decade following the discovery of the EPM2A and NHLRC1 genes. Hum Mutat. 2009;30:715-723.
18. Summers BA, Cummings JF, de Lahunta A. Veterinary Neuropathology. 1st ed. St. Louis, MO: Mosby: 1995:527.
19. Suzuki Y, Akiyama K, Suu S. Lafora-like inclusion bodies in the CNS of aged dogs. Acta Neuropathol. 1978;44:217-222.
20. Suzuki Y, Ohta K, Kamiya S, et al. Topographic distribution pattern of Lafora-like bodies in the spinal cord of some animals. Acta Neuropathol. 1980;49:159-161.