Signalment:
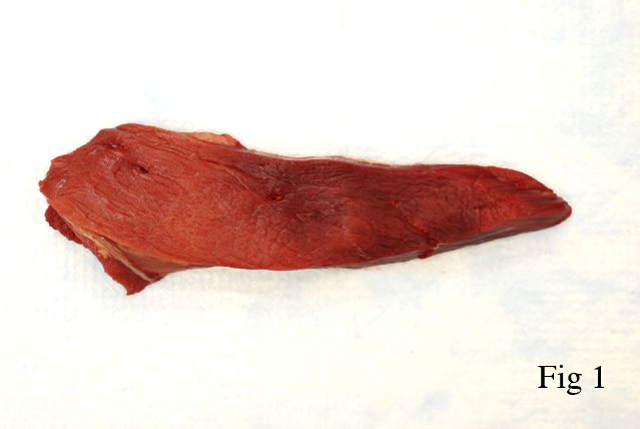
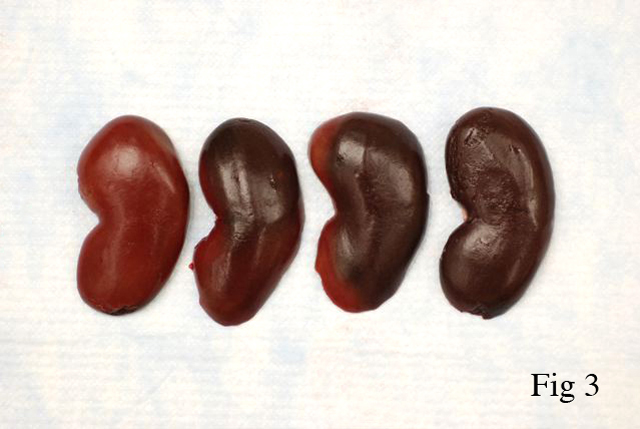
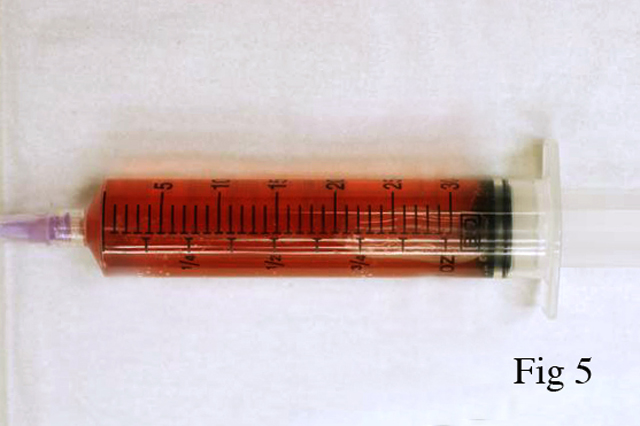
Gross Description:
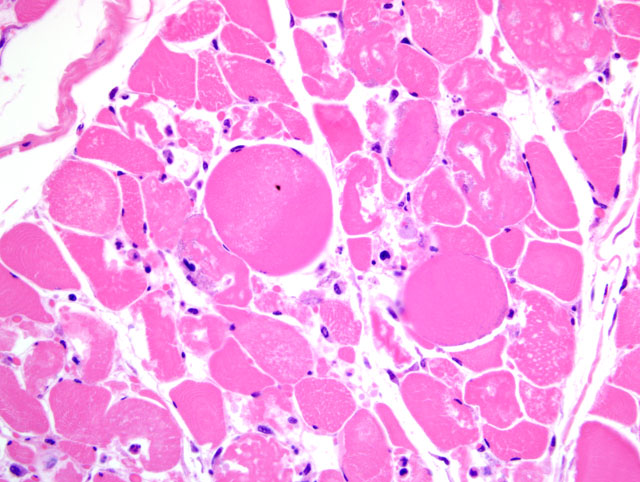
Histopathologic Description:
Morphologic Diagnosis:
Condition:
Contributor Comment:
Animals with capture myopathy may die suddenly or develop clinical signs hours, days or weeks later. Capture myopathy has been diagnosed up to a month after capture. Clinical syndromes of capture myopathy have been described based on time until onset of clinical signs as hyperacute, acute, subacute and chronic (reviewed in 4). More recently, other authors have based the classification of clinical syndromes on pathophysiology; capture shock syndrome, ataxic and myoglobinuric syndrome, ruptured muscle syndrome and a rare, poorly characterized delayed-peracute syndrome.(2) The ataxic and myoglobinuric syndrome is the most common, and is consistent with the present case.
Under normal conditions wild animals are not subjected to prolonged, maximal muscular exertion. However, during pursuit and capture such conditions may exist. Therefore, the earliest clinical signs of capture myopathy are similar to those of maximal exertion, increased respiratory and cardiac rates. Body temperature is usually elevated. Other early clinical signs may include depression, weakness, ataxia, muscle stiffness, and muscle tremors. Death may occur immediately post-capture due to marked metabolic acidosis, shock and circulatory collapse.
Animals surviving hours or even days may continue to show signs of depression, hyperthermia, tachypnea, tachycardia, weakness, and ataxia. Difficulty standing may progress to recumbency. Dark colored urine due to myoglobinuria may be seen. For weeks survivors may continue to show lameness, ataxia, muscle stiffness, and weight loss. Occasionally rupture of damaged muscle groups may occur. The gastrocnemius muscle is especially prone to rupture under such conditions.
The pathophysiology of capture myopathy is related to both shock and metabolic acidosis. The stress of pursuit and capture results in strong and prolonged sympathetic stimulation of microvasculature and eventual exhaustion of sympathetic vascular tone. Lack of vascular tone leads to visceral pooling of blood, decreased venous return, decreased cardiac output, hypotension, and hypoxia. In spite of hypoxia, tissue metabolism continues, relying on anaerobic glycolysis and resulting in increased levels of intracellular pyruvic and lactic acid. Lactic acid diffuses into the blood at levels that overwhelm the capacity of the liver, heart and other tissues to convert lactic acid to useable energy and lactic acidosis develops.
Prolonged hypoxia and acidosis result in generalized tissue deterioration. Active transport of sodium and potassium is reduced due to low intracellular pH. Intracellular sodium and chloride levels rise as do extracellular potassium levels. Mitochondrial activity decreases, lysosomes rupture, releasing damaging enzymes. Tissue necrosis ensues, especially in skeletal muscle, heart, liver and lung. Renal lesions of capture myopathy are characterized by moderate to severe tubular epithelial cell degeneration and necrosis with protein (myoglobin) and cellular casts. Renal lesions are primarily the result of renal ischemia.
Hyperthermia exacerbates tissue necrosis. Heat is generated from muscle myofilament action, glycolysis, recovery heat production as metabolic processes attempt to restore muscle to a resting equilibrium, and the environment. Heat from the environment can be transferred to muscle cells during exertion.(1)
Capture myopathy is similar to march myoglobinuria or extertional rhabdomyolysis in untrained athletes or military recruits following heavy exercise at high ambient temperatures.(2)
JPC Diagnosis:
Conference Comment:
Participants reviewed the basic stages of skeletal muscle necrosis, repair, and regeneration. Because myofibers are multinucleate, these changes can occur segmentally and are initially characterized by hyalinization of the sarcoplasm with loss of cross striations, followed by sarcoplasmic fragmentation often with mineralization. Effective skeletal muscle regeneration depends on the presence of an adequate blood supply, an intact basal lamina, and viable satellite cells. In the presence of adequate blood supply, macrophages derived from blood monocytes, with or without other leukocytes, are quickly recruited to the site of necrosis, traverse the basal lamina, and clear cytoplasmic debris. Simultaneously, satellite cells, juxtaposed between the sarcolemma and basal lamina, are activated and begin division into myoblasts in support of the regenerative effort. The remaining intact basal lamina forms a scaffold, i.e. sarcolemmal tube, which excludes fibroblasts and guides proliferating myoblasts, which then fuse end-to-end to form myotubes that eventually produce thick and thin filaments and mature into myofibers. By contrast, if large numbers of satellite cells are killed, even with persistence of the basal lamina, healing occurs by fibrosis rather than regeneration. In cases typified by loss or disruption of the basal lamina, even with persistence of viable satellite cells, regeneration is ineffective and healing is characterized by the formation of muscle giant cells (large, bizarre multinucleated giant cells) accompanied by fibrosis.(3)
References:
2. Spraker T: Stress and capture myopathy in artiodactylids. In: Zoo and Wildlife Medicine Current Therapy 3, ed. Fowler M, pp. 481-488. W.B. Saunders, Philadelphia, PA, 1993
3. Valentine BA, McGavin MD: Skeletal muscle. In: Pathologic Basis of Veterinary Diseases, ed. McGavin MD, Zachary JF, 4th ed., p9. 985-989, Mosby Elsevier, St. Louis, MO, 2007
4. Williams E, Thorne E: Exertional myopathy (capture myopathy ). In: Noninfectious Diseases of Wildlife, eds. Fairbrother A, Locke L, Hoff G, 2nd ed., pp. 181-193. Iowa State University Press, Ames, IA, 1996