Signalment:
Gross Description:
Histopathologic Description:
Morphologic Diagnosis:
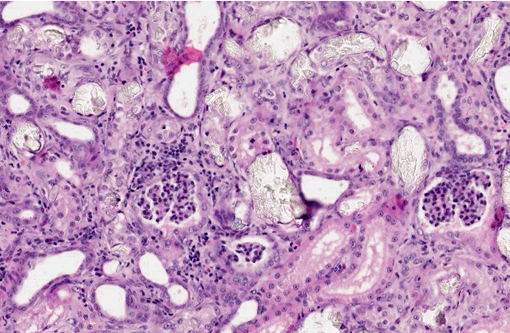
1. Kidney: Moderate, diffuse, subacute tubular necrosis and ectasia with multifocal tubular epithelial regeneration and many intratubular oxalate crystals.
2. Mild, multifocal lymphoplasmacytic and neutrophilic interstitial nephritis.
Condition:
Contributor Comment:
A naturally occurring disease in cats, clinically similar to PH2 in humans, has been described in several instances.(2,3,4) As with humans, the disease is autosomal recessive and typically affects kittens between five and nine months of age. Affected animals develop acute renal failure with increased urinary oxalate and L-glyceric acid levels. Histologically, abundant oxalate crystals are present within renal tubules and occasionally Bowmans spaces, accompanied by acute tubular necrosis and, variably, mild interstitial fibrosis.(3) Also consistent with human PH2, GRHPR mutation has been associated with the feline disease in cats from one colony.(4) A point mutation in the acceptor site of intron 4 was identified and correlated with a frameshift and premature stop codon in RNA transcripts from affected cats.
Several distinct differences exist in the clinical presentation of feline primary hyperoxaluria and PH2 in humans. Human PH2 is typically diagnosed in adults when renal changes are chronic; stone formation is usually less severe than that observed in PH1, which may present in infancy. By contrast, feline PH presents in young animals and is characterized by severe, acute disease. Concurrent neurologic lesions may accompany renal disease in cats and are histologically typified by swelling in the proximal axons of spinal motor neurons, ventral roots and intramuscular nerves and the dorsal root ganglia due to neurofilamentous accumulations. This lesion may be accompanied by Wallerian degeneration in peripheral nerves and associated denervation muscle atrophy. It is uncertain how these changes relate to the metabolic deficits present in primary hyperoxaluria or whether they represent a concurrent genetic defect.(3,7)
Primary hyperoxaluria must be distinguished from secondary disease due to exposure to large amounts of oxalates or increased absorption of dietary oxalic acid from the intestinal tract. In companion animals, acute oxalate nephrosis is typically seen in cases of ethylene glycol poisoning. In large animals, oxalate-rich plants are generally the source of toxicity.(8) Histologically, renal changes due to ethylene glycol toxicity are indiscernible from those present in cases of feline primary hyperoxaluria. A distinction between feline primary hyperoxaluria and oxalate toxicity is made on the basis of clinical history and supporting clinicopathological information (e.g. L-glyceric aciduria).
JPC Diagnosis:
1. Kidney: Tubular degeneration and necrosis, diffuse, marked, with regeneration, mineralization, and numerous intratubular oxalate crystals.
2. Kidney: Nephritis, interstitial, lymphoplasmacytic, multifocal to coalescing, mild, with fibrosis.
Conference Comment:
Finally, participants discussed toxic plants as the usual cause of oxalate nephrosis in ruminants. A partial listing of oxalate-containing plants implicated in such cases follows:(8)
| Scientific Name | Common Name |
| Halogeton glomeratus | Halogeton |
| Sarcobatus vermiculatus | Greasewood |
| Rheum rhaponticum | Rhubarb |
| Oxalis cernua | Soursob |
| Rumex spp. | Sorrel, dock |
References:
2. Danpure CJ, Jennings PR, Mistry J, Chalmers RA, McKerrell RE, Blakemore WF, et al. Enzymological characterization of a feline analogue of primary hyperoxaluria type 2: a model for the human disease. J Inherit Metab Dis. 1989;12:403-14.
3. De Lorenzi D, Bernardini M, Pumarola M. Primary hyperoxaluria (L-glyceric aciduria) in a cat. J Feline Med Surg. 2005;7:357-361.
4. Goldstein RE, Narala S, Sabet N, Goldstein O, McDonough SP. Primary hyperoxaluria in cats is caused by a mutation in the feline GRHPR gene. J Heredity. 2009;100:S2-S7.
5. G+â-+lbahar MY, Kaya A, G+â-¦len I. Renal Oxalosis in a Calf. Turk J Vet Anim Sci. 2002;26:1197-1200.
6. Hoppe B, Beck BB, Miller DS. The primary hyperoxalurias. Kidney Int. 2009;75:1264-1271.
7. Maxie MG, Youssef S. Nervous system. In: Maxie MG, ed. Jubb, Kennedy and Palmers Pathology of Domestic Animals. 5th ed. Philadelphia, PA: Elsevier Saunders; 2007:375.
8. Maxie MG, Newman SJ. Urinary system. In: Maxie MG, ed. Jubb, Kennedy and Palmers Pathology of Domestic Animals. 5th ed. Philadelphia, PA: Elsevier Saunders; 2007:468-473.
9. George JW, Zabolotzky SM. Water, electrolytes, and acid base. In: Latimer KS, ed. Duncan & Prasses Veterinary Laboratory Medicine Clinical Pathology. 5th ed. Ames, Iowa: Wiley-Blackwell; 2011:145-171, 430-432.