Signalment:
Gross Description:
Histopathologic Description:
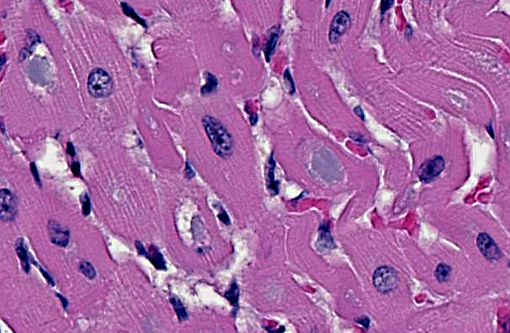
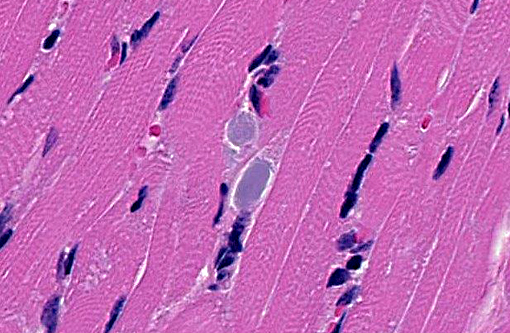
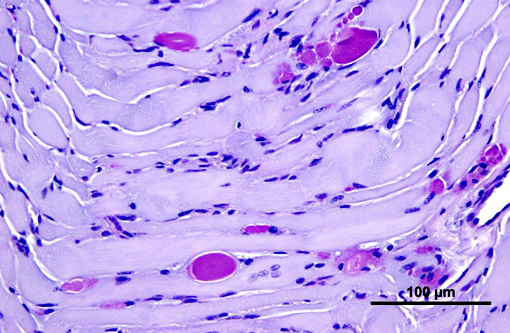
Heart: Intracytoplasmic inclusions, similar to those previously described, are within numerous variably swollen and disrupted myocytes.
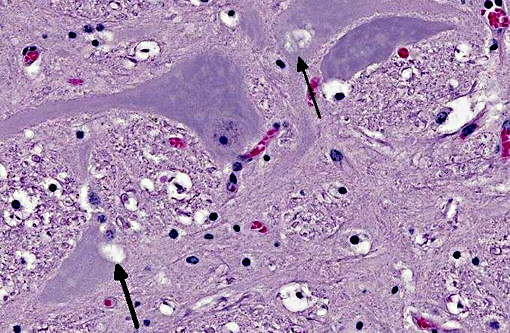
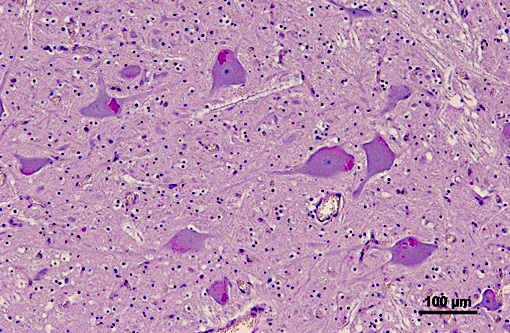
Brainstem: A few to moderate numbers of large neuronal cell bodies contain intracytoplasmic inclusion bodies, similar to those previously described.
Morphologic Diagnosis:
1. Heart, Skeletal muscle (diaphragm, intercostal, thigh): Severe multifocal myofiber degeneration with intracytoplasmic inclusions.
2. Brainstem: Moderate neuronal degeneration with intracytoplasmic inclusions.
Lab Results:
Genotyping was performed at the Veterinary Genetics Laboratory at the University of California, Davis. DNA used for analysis was isolated from formalin fixed paraffin embedded skeletal muscle tissue. Analysis showed the foal to be homozygous for the single nucleotide polymorphism responsible for glycogen branching enzyme deficiency.
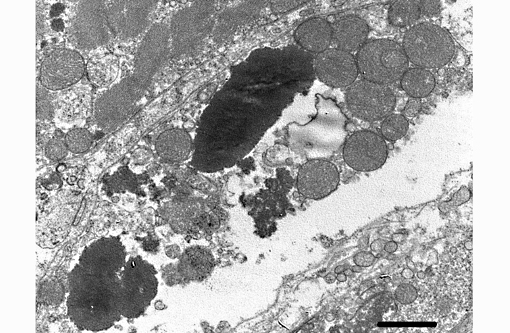
Transmission electron microscopy was performed on formalin fixed skeletal muscle and brainstem at the California Animal Health and Food Safety Laboratory, Davis Branch. Transmission electron microscopy revealed large electron dense filamentous to granular inclusions in degenerate skeletal muscle fibers. Similar inclusions are seen within degenerate neuronal cell bodies within the brainstem.
Condition:
Contributor Comment:
Normal glycogen is present in the cytosol in the form of small electron dense granules that range in diameter from 10 to 40 nm and are comprised of glucose molecules that have α 1, 4 linkages between glucose residues and α 1, 6 glycosidic bonds at every 10th residue that create a branched polymer.(3) Branching of the polymer increases water solubility and provides a larger number of terminal residues which are the sites for enzymatic action of glycogen phosphorylase and glycogen synthase; enzymes that are responsible for glycogen degradation and synthesis, respectively. While glycogen synthase catalyzes the synthesis of α-1, 4 linkages between glucose residues, glycogen branching enzyme is responsible for the formation of the α 1, 6 glycosidic bonds that create the branched form of the glucose polymer, glycogen. Animals born deficient of this enzyme form non-branching, PAS-positive, diastase-resistant glucose polymers that form large aggregates within the cytoplasm of various tissues including skeletal muscle, brain, spinal cord, heart, liver. Glucose polymers that have only α-1, 4 linkages between glucose residues or those having few α-1, 6 glycosidic bonds (1 per 30 glucose residues) are amylose and amylopectin, respectively.(3) Amylose and amylopectin are starches that serve as nutritional reservoirs for plants and are not normal storage forms of glucose in mammalian species. Glycogen branching enzyme deficiency (equine glycogen storage disease IV) has previously been termed amylopectinosis prior to identification of the genetic mutation responsible for this disease.(2) Also, prior to discovering the genetic mutation and inheritance pattern, Valberg, et al. reported that glycogen branching enzymatic activity was virtually absent in affected foals and that some of the half-siblings of the affected foals had an approximately 50% decrease in GBE activity.(6)
The breed of the foal (Quarter horse) and the character and staining properties of the numerous intracytoplasmic inclusions seen within all examined skeletal muscles, heart, and brainstem were consistent with glycogen branching enzyme deficiency, also referred to as glycogen storage disease IV. This diagnosis was confirmed by genotype analysis. The hemorrhages seen in multiple organs are considered agonal. The partial aeration of the lung parenchyma may have occurred during attempts at resuscitation or at the time of parturition when the foal was reported to have gasped for air prior to ceasing to breathe.
JPC Diagnosis:
1. Skeletal muscle: Glycogen-like inclusions, intrasarcoplasmic, many, with multifocal mild rhabdomyocyte degeneration.
2. Cardiac muscle: Glycogen-like inclusions, intrasarcoplasmic, many.
3. Brainstem, gray matter, neurons: Glycogen-like inclusions, intracytoplasmic, rare.
Conference Comment:
Glycogen is demonstrated histologically using the PAS reaction on two serial sections. Pretreatment of one section with diastase enables its comparison between the intensity of magenta staining of the two sections to determine whether glycogen is present. If glycogen is present, diastase will digest it and remove it from the tissue (diastase-sensitive), thus removing the magenta color. The inclusions in this case are diastase-resistant because, as the contributor describes, the deficient glycogen branching enzyme (GBE) is necessary to form the glycosidic bonds between glucose molecules, creating the branching polymer glycogen. This is in contrast to other glycogen storage diseases such as type I and III which involve deficiencies in the conversion of glycogen to glucose, thus resulting in excess accumulation of glycogen.(1) All types of glycogen storage diseases are characterized by defects in the glycogen metabolic pathway and ultimately result in the failure of adequate utilization of energy resources leading to generalized muscle weakness or death.
Another storage myopathy commonly recognized in the American Quarter horse, though reported in many other breeds, is equine polysaccharide storage myopathy. A definitive abnormality of the glycolytic or glycogenolytic pathway has not yet been identified for this entity, although a point mutation in skeletal muscle glycogen synthase I (GYS1) gene has been associated with some cases. This disease results in the accumulation of intracytoplasmic glycogen within type 2 fibers, which can become amylase-resistant inclusions as the glycogen becomes ubiquitinated in chronic cases. Episodes of exertional rhabdomyolysis is often associated with this condition.(5)
A second major group of storage diseases worthy of mention in domestic animals are those of lysosomes, characterized by a deficiency of lysosomal acid hydrolases leading to the excess accumulation of insoluble metabolites within lysosomes. For a case example and in-depth discussion of the numerous types of lysosomal storage diseases, we refer the reader to WSC 2013 (conference 5, case 2).
References:
1. Myers RK, McGavin MD, Zachary JF. Cellular adaptations, injury and death: Morphologic, biochemical, and genetic bases. In: Zachary JF, McGavin MD, eds. Pathologic Basis of Veterinary Disease. 5th edition. St. Louis, MO: Elsevier Mosby; 2012:35,54-55.
2. Render JA, et al. Amylopectinosis in a fetal and neonatal Quarter horse. Vet Pathol. 1999;36:157-160.
3. Stryer L. Biochemistry. 4th edition. New York: WH Freeman and Company; 1995:472-473, 581-588.
4. Valberg SJ, et al. Glycogen branching enzyme deficiency in Quarter horse foals. J Vet Intern Med. 2001;15:572- 580.
5. Valentine BA, McGavin MD. Skeletal muscle. In: Zachary JF, McGavin MD, eds. Pathologic Basis of Veterinary Disease. 5th edition. St. Louis, MO: Elsevier Mosby; 2012:902-904.
6. Wagner ML, et al. Allele frequency and likely impact of the glycogen branching enzyme deficiency gene in Quarter horse and Paint horse populations. J Vet Intern Med 2006;20:1207-1211.
7. Ward TL, et al. Glycogen branching enzyme (GBE1) mutation causing equine glycogen storage disease IV. Mamm Genome 2004;15:570-577.