Signalment:
The percutaneous abdominal ultrasonography revealed an enlargement in both kidneys with multiple cystic cavities filled with anechoic fluid. A larger cyst, measuring 6.7 cm in diameter, was found in the left kidney. The right kidney was enlarged but smaller than the left and also displayed multiple cysts. The animal presented a rapid decrease in its general condition due to renal failure, and the lack of improvement to initial fluid therapy indicated a poor prognosis and euthanasia was advised after five days. The necropsy was performed a few hours later in the same day.
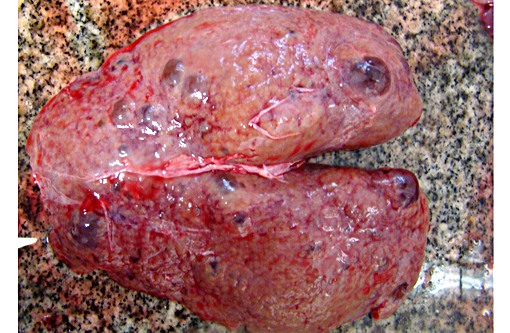
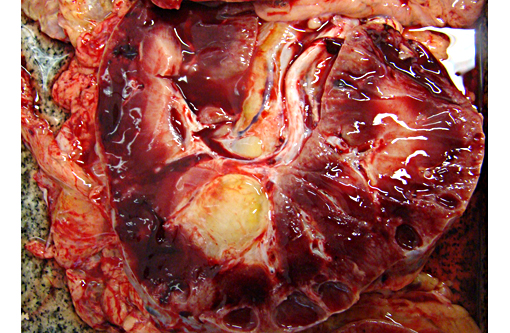
Gross Description:
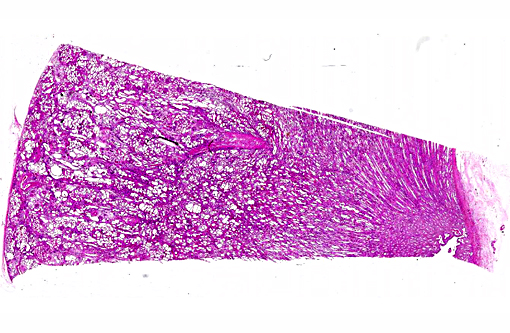
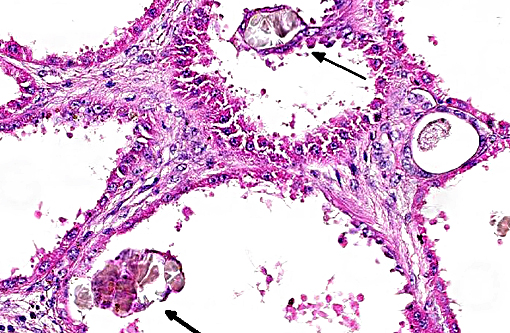
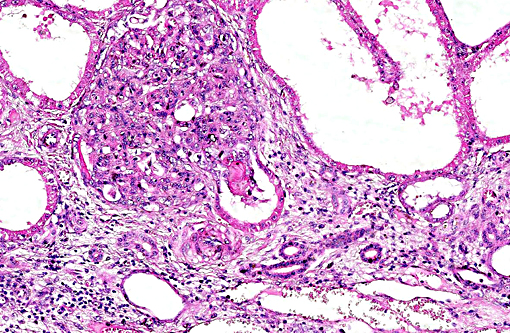
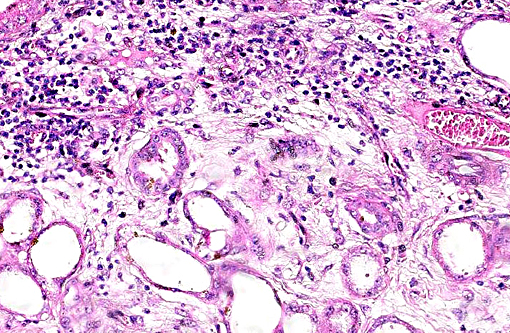
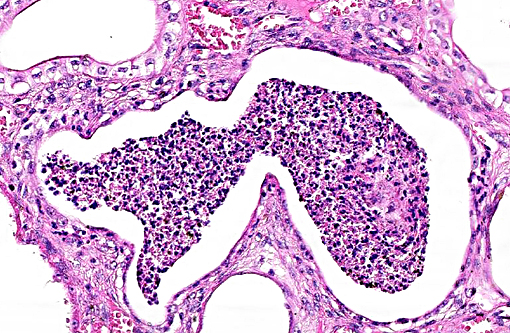
Histopathologic Description:
Morphologic Diagnosis:
Lab Results:
Hematology (ref. values):
RBC 6,7 x 106/mm³ (6.8 12,9 x 106/mm3)
Hematocrit 30 % (32-53%)
Hemoglobin 10.5 mg/ dL (11-19mg/dL)
MCV 44.78 fl
MCHC 35 %
WBC 15300 /mm³ (5400-14300 /mm³)
Neutrophils 89% (13.617) (2260-8580/ mm3)
Bands 0
Basophils 2% (306) (0-100/mm³)
Lymphocytes 7% (1071) (1500-7700/mm³)
Monocytes 0 (0-1000/mm³)
Eosinophils rare (0-800/ mm3)
Serum chemistry (ref. value):
Creatinine: 14,4 mg/dL (1.2-1.9 mg/dL)
BUN: 400 mg/dL (21.4-51.36 mg/dL)
Triglycerides: 807 mg/dL (22.9-46.0 mg/dL)
Cholesterol: 258.7 mg/dL (61-112 mg/dL)
Total protein: 6.4 mg/dL (5.2-7.9 g/dL)
Albumin: 2.9 mg/dL (2.653.69g/dL)
Ca e P: not measured
Abdominal ultrasonography: showed hyper-echoic, bilateral polycystic kidneys with the biggest cyst measuring 6.7 cm located in the left kidney cranial pole.
Urine culture (at necropsy): Klebisella pneum-oniae
Pericardial fluid (at necropsy): pH 7.5, specific gravity 1014, protein 1.2g/ dL, 200 nucleated cells/ mm3, without bacteria.
Abdominal fluid (at necropsy): pH7.5, specific gravity 1012, protein 0,8g/ dL, 600 nucleated cells/ mm3, without bacteria.
Condition:
Contributor Comment:
Cystic diseases of the kidney include various conditions characterized by one or more grossly visible cystic cavities in the renal parenchyma. Cysts can arise during organogenesis, and may be associated with histological criteria of renal dysplasia. In general, PKD is described as either 1) a congenital form or 2) an adult form. The congenital form is known to occur in dogs, cats, horses, cattle, sheep, pigs, several laboratory animal species, and humans. In humans, this congenital form is based on an autosomal recessive trait caused by mutation on the PKHD1 gene encoding fibrocystin, which is a receptor protein. It is proof that this genetic mutation follows an autosomal recessive trait similar to the childhood form of PKD in humans is also responsible for polycystic kidney disease in Persian kittens, lambs, and West Highland White and Cairn terrier puppies, whereas the inheritance of congenital PKD in other species is unknown.
Cysts can develop in any part of the nephron, including the glomerular space, or in the collecting system. In glomerulocystic disease, they only develop in Bowman's space, but this exclusivity is exceptional. There is no evidence that cysts are caused by a failure of nephrons to unite with the collecting system. Analysis of their content indicates that they are part of functional nephrons, and that their activity is consistent with their location in the nephron. Three mechanisms, which are not mutually exclusive, may lead to the formation of renal cysts. The pathogenesis of cyst formation in PCKD is thought to represent a combination of altered cell growth, fluid secretion and altered composition of the extracellular matrix.
Nevertheless, the gross and histopathological similarities of the lesions together with the progressive nature of lesions resulting in presentation in middle to old age is similar to these entities, implying a similar pathogenesis. As such, it would appear most likely that equine PCKD results from a random genetic defect resulting in downstream malformation of distal tubules/collecting ducts within the kidneys, ultimately culminating in effacement of normal renal architecture by progressively enlarging space occupying fluid-filled cysts. Renal cysts can be subdivided into congenital or acquired lesions. Solitary renal cysts occur in many species, most commonly in pigs and cattle, and are essentially incidental findings. Renal cysts become clinically significant when multiple and bilateral; such cases are named polycystic kidneys. Polycystic kidney disease (PCKD) is best characterized in man with further subdivision into autosomal dominant and autosomal recessive forms.
Autosomal recessive PCKD is rare and usually is associated with disease in childhood that is often severe. Those patients who do survive may progress to the development of hepatic fibrosis. In veterinary species, a similar entity has been described in Cairn terriers, characterized by juvenile onset and multiple cysts in both kidneys and liver. Polycystic kidneys are uncommon in horses; to our knowledge, there have been only five previous cases of PCKD in mature horses. In those cases, clinical signs (severe weight loss and anorexia) were similar to our case, although some author had related hematuria as the main clinical sign. Both kidneys were presented enlarged, containing multiple cystic structures that distorts the normal renal architecture are the classical gross findings.
Studies in man and Persian cats have been carried out in attempt to identify the segment of the nephron which is involved in cyst development. In man, the cysts have been identified as focal dilations of proximal or distal convoluted tubules of the nephron, with occasional involvement of glomeruli. The cysts result from initial epithelial proliferation which then becomes separated from the tubule lumen and is filled not only by glomerular filtrate but also by a dysfunction of epithelial fluid absorption and secretion.
With regard to hematological and biochemical parameters, major biochemical abnormalities include consistently increased urea and creatinine levels, as would be expected with a chronic renal disease resulting in progressive reduction of renal function. Anemia was reported in cases where hematuria was a clinical finding but not in the present one and in those who this clinical sing was absent; therefore, this abnormality may reflect blood loss rather than diminution of erythropoietin production by the peritubular interstitial tissue of the kidney. Although secondary hyperparathyroidism due to chronic renal failure is a common sign, it was not found in the present case.
Despite the morphological similarity of the lesion seen in man and Persian cats, it is unlikely that a similar familial genetic defect is responsible for the disease in horses, given the paucity of reported cases and lack of evidence of familial association disease. Nevertheless, the gross and histopathological similarity between lesions together with the progressive nature of lesions resulting in presentation in middle to old age is similar to these entities, implying a similar pathogenesis. As such, it would appear most likely that equine PCKD results from a random genetic defect resulting in downstream malformation of distal tubules/collecting ducts within the kidneys, ultimately culminating in effacement of normal renal architecture by progressively enlarging space occupying fluid-filled cysts. Immunohistochemistry can be performed in order to clarify the segment of epithelium in cyst formation. A combination of vimentin and a specific anticytokeratin antibody direct against cytokeratin 18 showed to be useful to characterized collect tubule involvement.
JPC Diagnosis:
Conference Comment:
This case was studied by Dr. Rachel Cianciolo at the Ohio State University, who has extensive experience and a keen interest in veterinary nephropathology; she interprets the renal changes as representing two simultaneous ongo-ing processes: 1) obstructive nephropathy with tubular dilatation and hyperplastic renal pelvis epithelium, which may be related to the evidence of urolithiasis described grossly, and the presence of oxalate crystals in the tubules; and 2) proliferative and sclerosing glomerulonephro-pathy, which may or may not be immune complex-mediated. She also observes that there is protracted tubular epithelial degeneration and necrosis with secondary interstitial edema and inflammation that could be due to either of the above processes. She further elaborates that polycystic kidney disease, as it occurs in human medicine, specifically refers to hereditary syndromes, either autosomal dominant or autosomal recessive; polycystic kidney is most appropriately applied when the specific genetic mutation has been identified or there is support for a heritable process. In this case, the tubular dilation, degeneration and necrosis are likely related to urolithiasis / crystalluria, and which is aligned more closely with an acquired cystic disease.
This case was additionally studied in consultation with the Departments of Genitourinary and Nephropathology at the Joint Pathology Center, whose medical pathologists are familiar with polycystic kidney disease as described in humans. They indicated the renal lesions in this case are not consistent with polycystic kidney disease, at least as it occurs in humans. Similar to conference participants, the medical pathologists identified dilated tubules with atrophic tubular epithelium, including within collection ducts, and a mesangioproliferative glomerulonephritis with decreased numbers of glomeruli. They characterize the lesion as chronic tubulointerstitial disease, and also describe arteriolar and arterial sclerosis. They also commented on the birefringent, smooth-surfaced crystals within tubules and within the interstitium and stated that, by light microscopy, the crystals are consistent with calcium oxalate. They also mentioned the presence of basophilic non-birefringent crystals vicinity the renal pelvis, consistent with calcium phosphate.
The underlying cause of the glomerular changes remains uncertain in this case. Typically, glo-merulonephritis is described as an immune meditated disease and the proliferative (or me-sangioproliferative) form occurs most commonly in horses. Glomeruli can also be non-specifically involved in association with other renal condi-tions, such as tubulointerstitial disease,(4) which may have caused the glomerular changes in this case vice an immune mediated process. Other non-immune mediated causes of glomerular injury include hypertension and coagulation secondary to endothelial injury.(4)
That said, the glomerular lesions in this case were severe enough to result in crescent formation, a finding which generally occurs as a response to fibrin exudation into the urinary space, followed by invasion of mononuclear cells, proliferation of parietal epithelium, and production of collagen by fibroblasts.(4) Additionally, the presence of synechia, which occurs with loss of podocytes and adhesion of the glomerular basement membrane to the parietal epithelium, is another indicator of severe glomerular damage. Glomerulonephritis in horses is apparently not uncommon, but progression to renal failure is reported as rare. Infectious causes in horses include equine infectious anemia and Streptococcus equip. The contributors necropsy observations of a urinary bladder with thickened, irregular red mucosa and reported urine culture of Klebisella pneumonia provide further plausible explanation for acquired renal tubular disease with simultaneous glomerular lesions.
References:
1. Aguilera-Tejero E., Estepa J.C., L³pez I., Bas S, Rodriguez M. Polycystic kidneys as a cause of chronic renal failure and secondary hypoparathyroidism in a horse. Equine Vet J. 2000;32:167-169.
2. Biller DS, DiBartola SP, Eaton KA, Pflueger S, Wellman ML, Radin MJ Inheritance of Polycystic Kidney Disease in Persian Cats. J Hered. 1996;87:l-5.
3. Cowley BD, Gudapaty S, Kraybill AL, Barash BD, Harding M A, Calvet, J P, Gattone VH. Autosomal-dominant polycystic kidney disease in the rat. Kidney Int. 1993;43: 522-534.
4. Maxie MG, Newman SJ. Urinary System. In: Maxie MG ed. Jubb, Kennedy, and Palmer's Pathology of Domestic Animals. 5th ed. Vol 2. Philadelphia, PA: Elsevier Saunders; 2007:442-462.
5. Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:23842398.
6. Iverson W.O., Fetterman,G.H., Jacobson E.R., Olsen J.H., Senior D.F., Schobert EE. Polycystic kidney and liver disease in Springbok. 1. Morphology of the lesions. Kidney Int. 1982; 22:146155.
7. Muller DWH, Szentiks CA, Wibbelt G. Polycystic Kidney Disease in Adult Brazilian Agoutis (Dasyprocta leporina). Vet Pathol. 2009;46:656661.
8. Plesker R., Schulze H. Polycystic nephropathy in slender lorises (Loris lydekkerianus). Am J Primatol. 2006;68:838844.
9. Rhind SM, Keen JA. Polycystic kidney disease in a mature horse: report and review of previously reported cases. Equine Vet Educ. 2004;16:178-183.
10. Takahashi H, Calvet J P, Dittemore-Hoover D, Yoshida K, Grantham JJ, Gattone VHA. Hereditary Model of Slowly Progressive Polycystic Kidney Disease in the Mouse. J Am Soc Nephrol. 1991;1:980-989.
11. Torre VE, Harris PC. Mechanisms of disease: autosomal dominant and recessive polycystic kidney diseases. Nat Clin Pract Nephrol. 2006;2(1):40-55.