Signalment:
Gross Description:
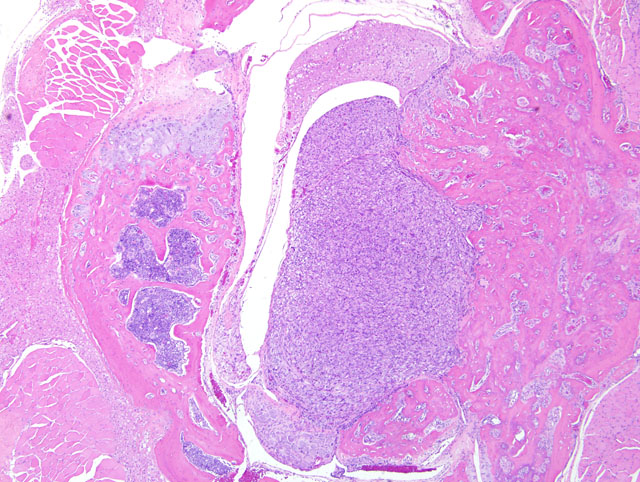
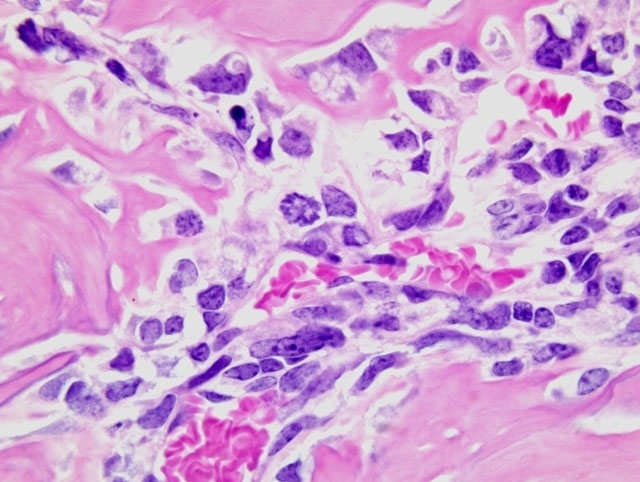
Histopathologic Description:
Morphologic Diagnosis:
Condition:
Contributor Comment:
This is a typical osteoblastic osteosarcoma with abundant osteoid production making diagnosis fairly simple. Osteosarcomas in mice can be osteoplastic, chondroblastic, osteoclastic, anaplastic, osteoblastic, fibroblastic, telangiectatic (vascular), or compound.(3) The National Toxicology Program records an overall incidence of osteosarcomas in 9-month-old p53 transgenic mice of 2.0% in males and 7.3% in females.(2) In the study in question, the maxilla was the most common site of osteosarcomas, with the vertebrae second most common; osteosarcomas were also recorded in the femur, tibia, and rib. In a mouse model of osteosarcoma development, wherein one or both p53 alleles were inactivated with or without inactivation of one or both Rb alleles, the most common sites of osteosarcoma development were the jaw (mandible followed by maxilla), followed by the hind leg/ hip, and ribs and vertebrae.(6) Metastases were present (largely in the lungs and liver) in 9% of mice, and rarely in those animals presenting with tumors on the jaws (presumably because they had a significantly decreased lifespan due to the primary tumor). It was noted in that study also that many tumors were microscopic.(6)
The incidences of several tumors, predominantly osteosarcomas and soft tissue sarcomas, are increased in p53 transgenic mice.(1) This observation, together with the observations that a high percentage of osteogenic sarcomas have rearrangements or deletions of the p53 gene and that bone tumors occur as part of a syndrome of germline mutations of p53 (the Li-Fraumeni family syndrome), supports the hypothesis that inactivation of p53 is an important step in development of bone tumors.(1)
JPC Diagnosis:
Conference Comment:
As indicated by its distinction as the guardian of the genome and molecular policeman, p53 is among the most influential and best characterized tumor suppressor genes known, and is the most common target for genetic damage in human neoplasia; p53 mutations are identified in over 50% of human tumors. The p53 gene encodes the p53 protein, a transcription factor whose role is essentially to prevent the propagation of cells with genetic damage, a function it accomplishes via three mechanisms: 1) temporary cell cycle arrest (i.e. quiescence), 2) permanent cell cycle arrest (i.e. senescence), and/or 3) apoptosis. Cell cycle arrest conferred by p53 is largely mediated by p53- dependent transcription of the CDK inhibitor p21, which inhibits cyclin-CDK complexes and phosphorylation (i.e. inactivation) of the retinoblastoma (RB) protein; the inhibition prevents cell cycle progression from the late G1 phase to the S phase (i.e. the point of no return) at the G1/S checkpoint, providing an opportunity for repair of DNA damage. Apoptosis, on the other hand, is induced by p53 in the face of irreversible DNA damage, and results from p53-directed transcription of such pro-apoptotic genes as BAX and PUMA.(5)
The contributor alluded to the germline p53 mutation in this transgenic mouse as analogous to the Li-Fraumeni syndrome of humans, discussion of which is best preceded by a cursory review of the two-hit hypothesis of oncogenesis. This hypothesis applies to a number of genes, but best illustrated by the RB tumor suppressor gene. Basically, the two-hit hypothesis states that oncogenesis depends on the presence of two mutations (i.e. hits) involving both alleles of a given tumor suppressor gene. As applied to the RB gene, retinoblastoma occurs only in the presence of two mutations one in each allele. Children with familial retinoblastoma inherit one defective copy of the RB gene as the result of a germline mutation (i.e. the first hit), and the second (i.e. somatic) mutation occurs sporadically. In sporadic cases, a single retinoblast must suffer two separate somatic mutations in the RB gene one in each allele to result in retinoblastoma. Therefore, children who inherit a germline RB mutation are at increased risk for developing retinoblastoma, because only one additional hit is required to confer complete loss of RB function. A germline mutation in p53 is the basis of the Li-Fraumeni syndrome, which is directly analogous to germline RB mutations in familial retinoblastoma. Affected humans with the Li-Fraumeni syndrome have a 25-fold higher chance of developing malignant neoplasia by age 50 than the general population.(5)
Wide histomorphological variation within a given tumor and between individual cases is typical of osteosarcoma, which may complicate tumor classification.(4) Accordingly, the neoplasm in this case exhibits substantial intratumoral histomorphologic variation, ranging from an expansile highly productive zone at the periphery, to an invasive lytic internal zone. Based upon this finding, the conference moderator encouraged participants to consider and discuss alternative diagnoses, including osteosarcoma arising in osteoma, or even a collision tumor. Close examination of the interface between the neoplasm and surrounding muscle is helpful in excluding these possibilities; in osteoma and reactive bone, as alluded to in case I above, a gradual transition between a dense fibrous layer and a well-differentiated layer of osteoblasts lining trabeculae is expected, whereas no such transition is present in this case and atypical ovoid neoplastic cells are present throughout the neoplasm.
References:
2. Kissling GE: Historical control rates for NTP Tg.AC p53 mice. National Toxicology Program (available at http://ntp.niehs.nih.gov/?objectid=522CD99F-F1F6-975E-74484178722ECB7C), 2008
3. Long PH, Leininger JR: Bones, joints, and synovia. In: Pathology of the Mouse, ed. Maronpot RR, pp. 665-671. Cache River Press, Vienna, IL, 1999
4. Slayter MV, Boosinger TR, Inskeep W, Pool RR, D+�-�mmrich K, Larsen S: Histological Classification of Bone and Joint Tumors of Domestic Animals, 2nd series, vol. I, ed. Schulman FY, pp. 9-11. Armed Forces Institute of Pathology (in cooperation with the CL Davis DVM Foundation and The World Health Organization Collaborating Center for Comparative Oncology), Washington, DC, 1994
5. Stricker TP, Kumar V: Neoplasia. In: Robbins and Cotran Pathologic Basis of Disease, eds. Kumar V, Abbas AK, Fausto N, Aster JC, 8th ed., pp. 273-292. Saunders Elsevier, Philadelphia, PA, 2010
6. Walkley CR, Qudsi R, Sankaran VG, Perry JA, Gostissa M, Roth SI, Rodda SJ, Snay E, Dunning P, Fahey FH, Alt FW, McMahon AP, Orkin SH: Conditional mouse osteosarcoma, dependent on p53 loss and potentiated by loss of Rb, mimics the human disease. Genes Dev 22:1662-1676, 2008