WSC 2022-2023
Conference 24
Case IV:
Signalment:
19 months old, female, mixed-breed, Sus scrofa domesticus, domestic pig.
History:
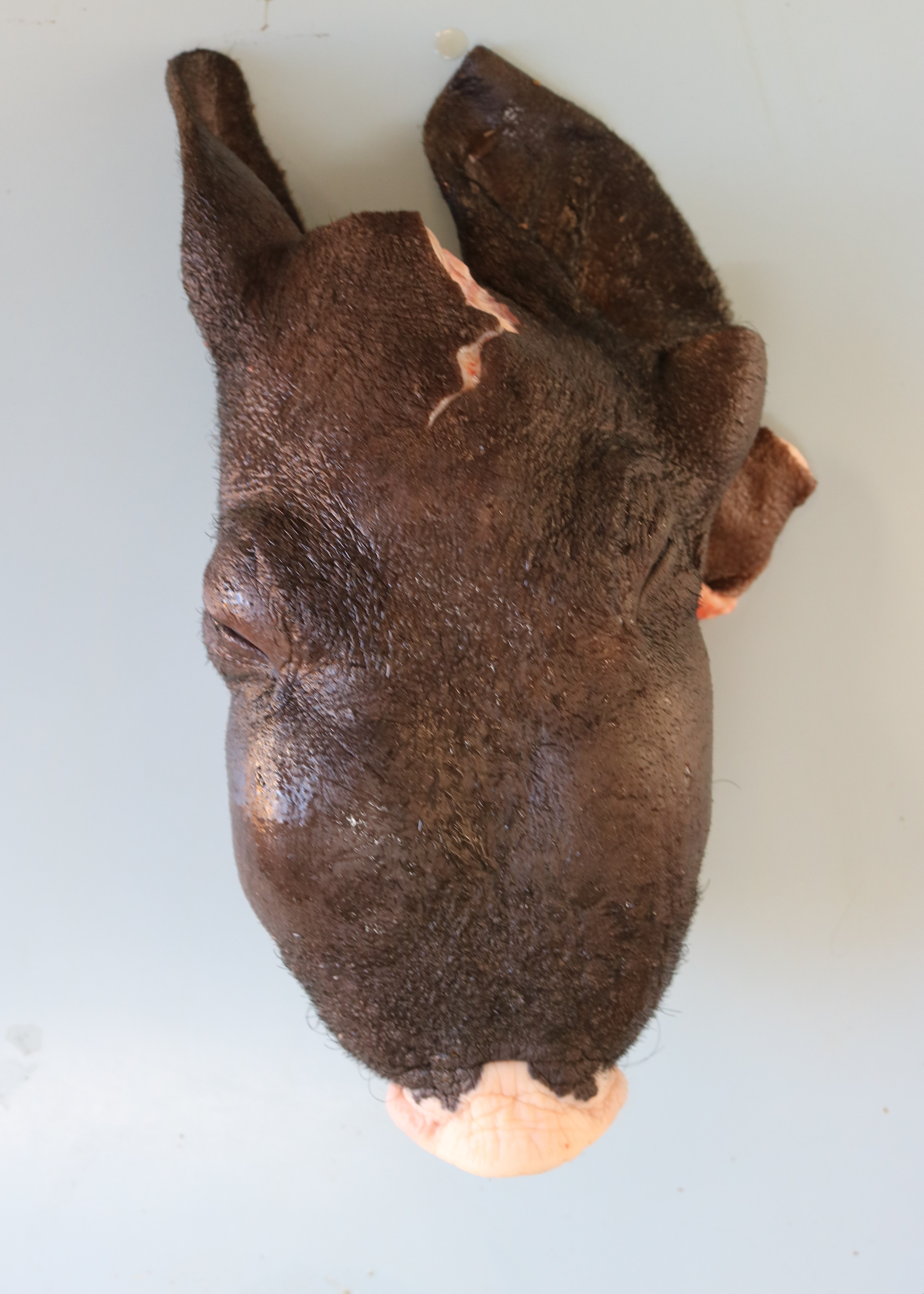
The pig was from a small farm housing pigs and other animals, including one boar and four sows that farrowed 23 piglets. After birth, the piglets were divided into four pens containing piglets of the same size from different sows. The number of piglets in each pen were as follows: pen 1, 8 piglets; pen 2, 4 piglets; pen 3, 9 piglets; and pen 4, 2 piglets. All pigs were fed a diet of boiled corn bran and millet; however, only the piglets in pen 3 became ill. At 10 months old, the first clinical signs appeared in the piglets, and their condition progressively worsened. Initially, the piglets presented with respiratory distress and underdevelopment. Later, they developed bilateral swollen face, mainly in the maxilla (Fig. 1), partial mouth opening and protruding tongue (Fig. 2), difficulty in food retention and mastication, loss of general body condition, and dyspnea. Subsequently, all pigs in pen 3 died under these conditions.
Gross Pathology:
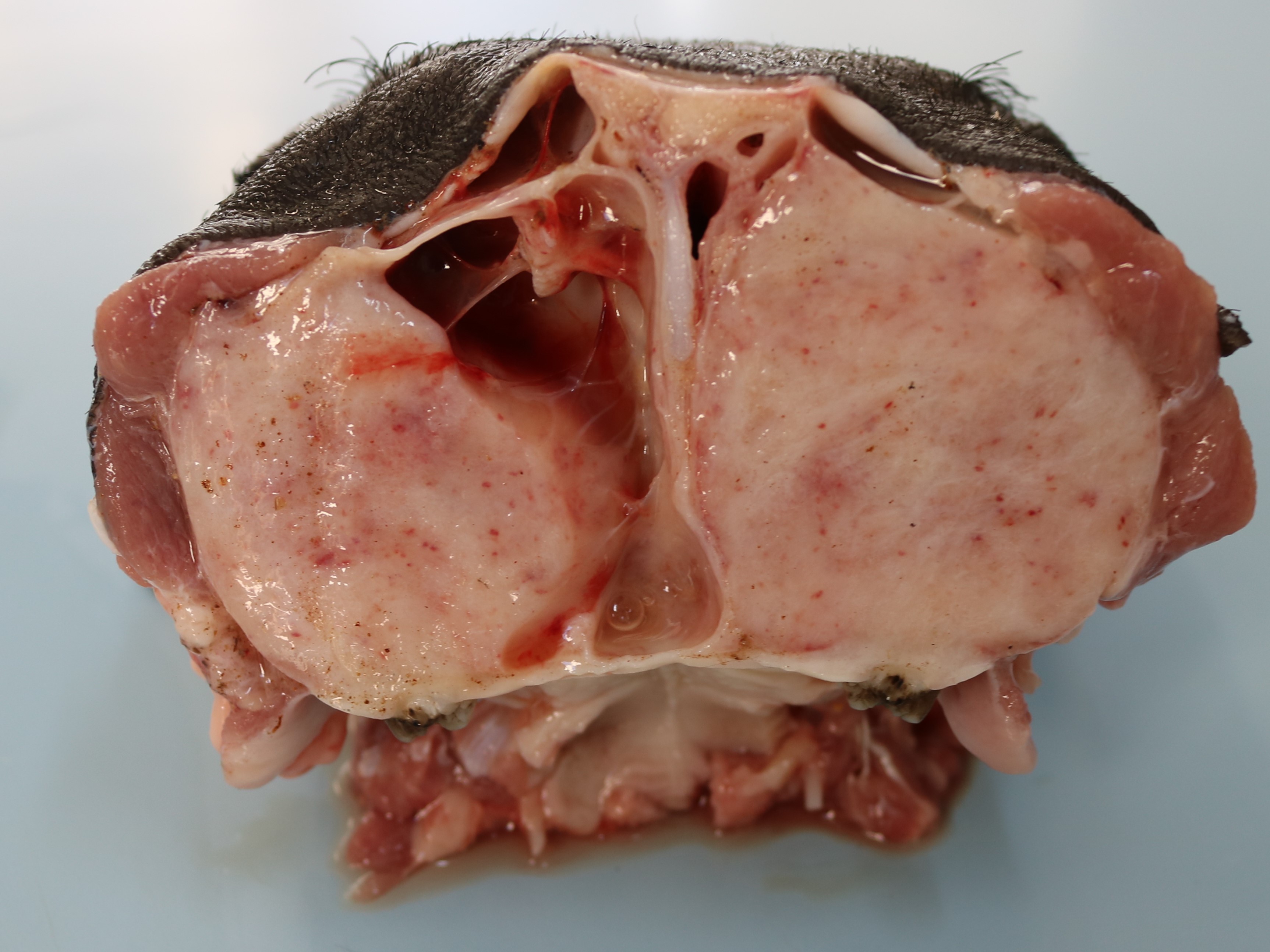
One piglet was euthanized and underwent necropsy. Marked bilateral enlargement of the facial bones, predominantly in the maxilla, was observed. These were easily sliced using a knife and the section surfaces were observed to be pale. The nasal cavity lumen decreased due to thickening of the maxillary bones. The teeth were loose, and the molars were lingually tipped. In addition, the ribs were soft and broke effortlessly under pressure. Most of the long bones showed epiphyseal plate thickening, and the trabecular structure was poorly visible.
Laboratory Results: None
Microscopic Description:
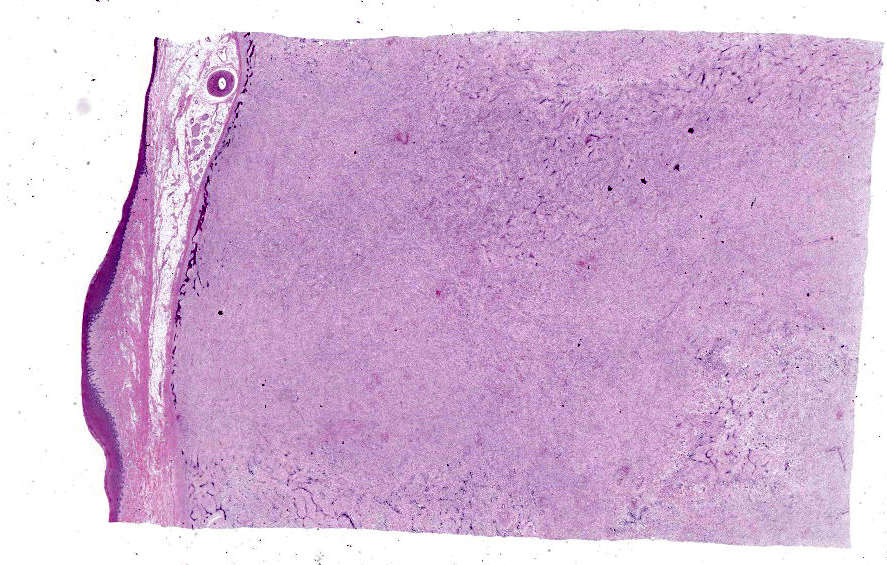
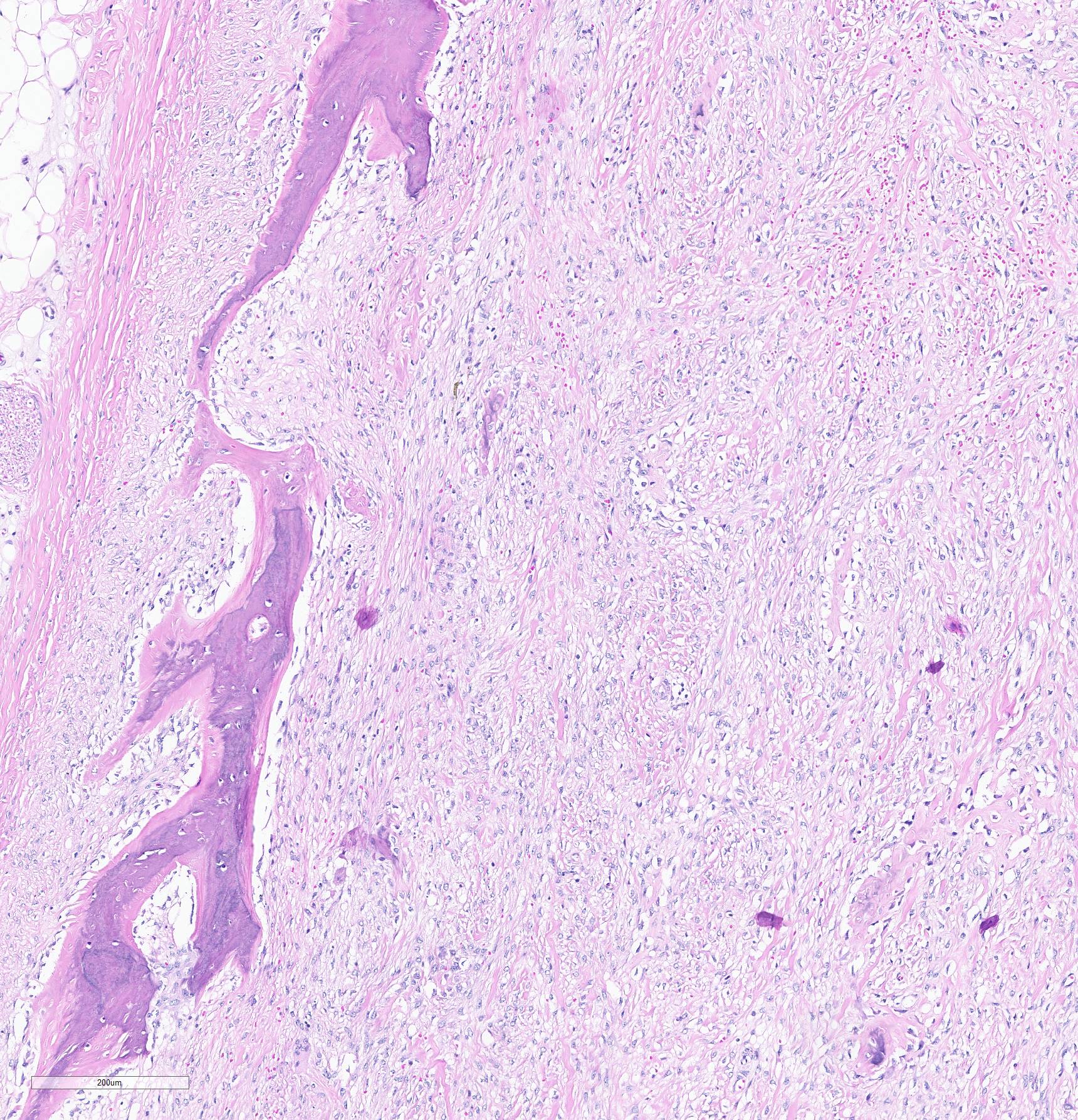
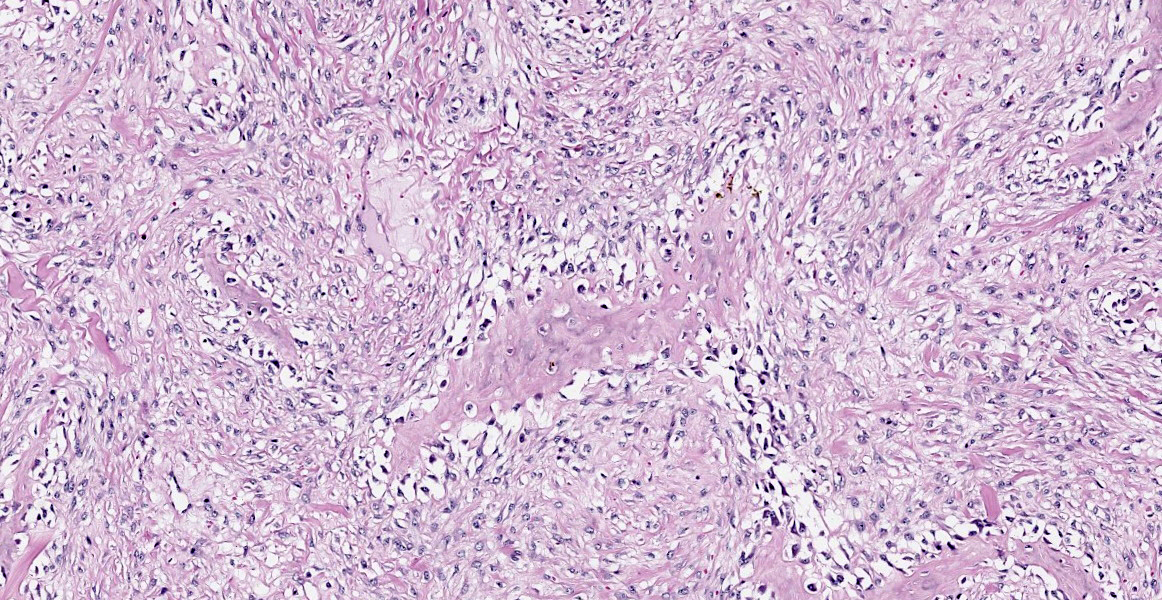
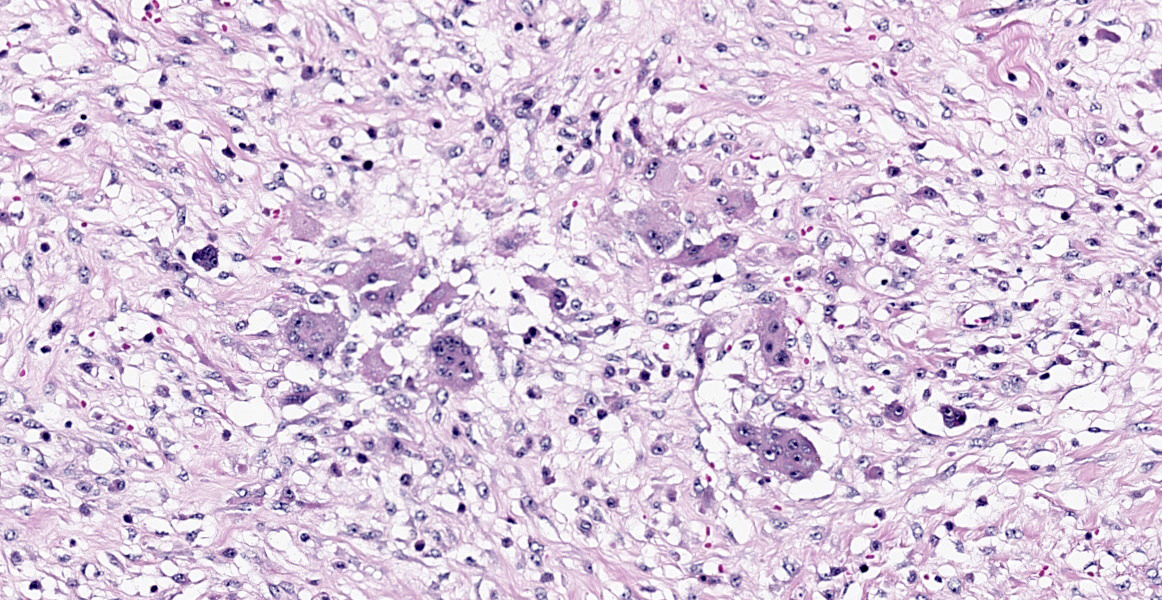
Histopathological examination of the mandible and maxilla revealed accentuated
diffuse proliferation of soft and irregular fibrous conjunctive tissue, which was evaluated by Masson’s trichrome staining around the osteoid trabeculae, many of which were partially or completely demineralized, as observed on von Kossa staining. Osteoclasts forming multinucleated cells were observed in the surface grooves (Howship’s lacunae) in the mineralized trabeculae or clustered within the connective tissue. Histological changes were not observed in the renal parenchyma or parathyroid gland.
Contributor’s Morphologic Diagnoses:
Face bones, maxilla: osteolysis, diffuse, severe fibroplasia (fibrous osteodystrophy), domestic pig (Sus scrofa domesticus)
Contributor’s Comment:
Bone growth and maturation are complex processes involving an interaction between genetic factors, local and systemic hormones, dietary nutrients, and mechanical forces. Any factors interfering with the synthesis of proteoglycans or collagen by chondroblasts or osteoblasts, differentiation of precursor cells, or resorption of bone by osteoclasts can result in skeletal abnormalities. The expression of an abnormality depends on many factors, including the phase of skeletal development that is altered, severity of the defect, age of the animal at the time of insult, and persistence of the influencing factor. Thus, the range of possible skeletal defects is large and a single cause can develop several different manifestations5.
Understanding the regulation of phosphorus and calcium levels in the animal body is necessary to comprehend the pathogenesis of bone lesions. Most mediators involved in calcium phosphorus homeostasis have evolved in fish, although their roles are repurposed during adaptation to land environments. Thus, while susceptibility to vitamin D deficiency in vertebrates can be traced back to fish, the specifics of its manifestation as a disease show both similarities and differences to what is observed in humans. Calcium and phosphorus are strictly regulated in all animals because of their vital roles in numerous cellular functions. During the colonization of land, vertebrates transitioned from a sea environment that was comparatively richer in calcium than phosphorus to a land environment, with a six- fold higher gravity force, that was poor in calcium and relatively richer in phosphorus. This led to the evolutionary changes involving major adaptations in bone structure and calcium/phosphorus regulation. These adaptations included a highly efficient system for intestinal calcium absorption and flexible bone remodeling, which allowed the use of bones as an internal reservoir of calcium to compensate for variations in dietary intake14.
Deficiencies in calcium, phosphorus, and vitamin D disrupt bone conditions, resulting in a group of diseases known as osteodystrophies or metabolic bone disease. In the field of veterinary medicine, this is a broad term denoting pathological conditions affecting multiple bones. Osteodystrophies are most frequently caused by genetic, nutritional, and/or hormonal abnormalities that affect bone growth, modeling, or remodeling, typically through disruptions in calcium/phosphorus metabolism. Calcium and phosphorus are required for a variety of essential bodily functions in addition to those involved in skeletal development. The skeleton is composed of approximately 99% of calcium and 85% of phosphorus in the body. There are three distinct forms of calcium and phosphorus in the body: ionized, protein-bound, and complex atoms in extracellular fluids (including plasma). The ionized fractions of these minerals (Ca2+ and HPO42-) are physiologically active and are strictly regulated by the parathyroid hormone (PTH), 1,25- dihydroxyvitamin D (1,25[OH]2D3), calcitonin, and phosphatonin system5,6,9,14.
PTHs are secreted by the parathyroid gland when calcium-sensing receptors on chief cells detect low serum ionized calcium concentrations. The net effect of PTH increases plasma ionized calcium levels and reduces plasma phosphate concentrations. The PTH induces the renal tubules to open the calcium channel, allowing increased resorption of calcium from the renal filtrate and increased breakdown of phosphate channels. Consequently, the resorption of phosphorus from the renal filtrate decreases. In the bone, high PTH concentration results in the recruitment and activation of osteoclasts, leading to increased osteoclastic bone resorption and release of calcium and phosphorus in the circulation5.
Vitamin D exists in two forms: vitamin D2 (ergocalciferol), obtained from yeasts and plants and vitamin D3 (cholecalciferol), obtained from the diet or as an end product of the skin photochemical reaction of 7-dehydrocholesterol, which explains why sun exposure increases vitamin D levels in most species. Latitude, time of day, season, and level of skin pigmentation can affect the vitamin D3 production in the skin. At lower angles, the sun does not have the requisite intensity to produce vitamin D in the skin. Furthermore, high levels of melanin in the skin absorb ultraviolet photons, making it unavailable for vitamin D synthesis. A dense hair/wool coat also reduces cutaneous vitamin D3 synthesis. Dogs and cats are an exception in mammals in that they do not produce vitamin D in the skin, owing to the presence of an enzyme that breaks down 7-dehydrocholesterol, making it unavailable for conversion to vitamin D. Therefore, cats and dogs are reliant on dietary vitamin D.
Vitamin D2 from the diet and vitamin D3 from the skin/diet are transported to the liver, where they form 25-hydroxyvitamin D (25OHD), the major form of vitamin D in the circulation. Therefore, serum 25OHD concentration reflects the level of cutaneous vitamin D3 formation and/or dietary levels of vitamin D. Serum 25OHD levels are measured to determine whether an individual has adequate or deficient vitamin D status. The most active form of vitamin D is 1,25-dihydroxyvitamin D, which is formed in the renal proximal tubular epithelial cells. This step is closely regulated and 1,25(OH)2D3 production is directly stimulated by high PTH and low phosphorus levels, and indirectly by low ionized calcium, which acts via PTH. When plasma phosphorus and ionized calcium concentrations are adequate and PTH levels are low, 25OHD is converted to inert metabolites during the initial step of the degradation pathways. Active vitamin D binds to the nucleus of the renal tubular and intestinal epithelial cells, resulting in increased absorption of calcium and phosphorus from the kidneys and intestine, inhibition of PTH production in the parathyroid gland, and negative feedback to decrease its own production5,6,14.
Calcitonin is secreted by thyroidal C cells in response to increased serum ionized calcium concentrations. It inhibits osteoclast action leading to decreased osteoclastic bone resorption, and subsequent decrease in the release of calcium and phosphorus into the blood, allowing normalization of serum ionized calcium levels5.
Recently, many phosphatonins have been found to be involved in phosphorus metabolism, the most important of which is the fibroblast growth factor 23 (FGF23). FGF23 is produced by osteocytes in the bone and its production is increased by either hyperphosphatemia or increased 1,25(OH)2D3. The kidney is the main target organ of FGF23; together with its cofactor klotho, FGF23 downregulates phosphorus channels in the kidney, resulting in decreased resorption of phosphorus from the renal tubules. FGF23 also decreases the production of 1,25(OH)2D3 by inhibiting the enzyme that acts in its formation and activates the enzyme that catabolizes 1,25(OH)2D3. in the parathyroid gland, FGF23 decreases secretion of PTH. FGF23 activity results in decreased plasma phosphorus concentration5.
Metabolic bone diseases can occur when these regulatory mechanisms do not work in harmony. Metabolic bone diseases include rickets, osteomalacia, fibrous osteodystrophy, or osteoporosis; these distinct morphological entities have characteristic pathogenesis and lesions. Nonetheless, specific diagnosis is difficult in many cases, as multiple conditions may be present, especially in those induced by nutritional deficiencies. This means that cases reported in the literature should be scrutinized, and only those confirmed by histopathology should be considered definitive5,6,14.
Fibrous osteodystrophy (osteodystrophia fibrosa, osteitis fibrosa cystica) is a relatively common metabolic bone disease characterized by extensive osteolysis, accompanied by proliferation of fibrous tissue and poor mineralization of the immature bone. The pathogenesis involves primary or secondary hyperparathyroidism with persistently elevated plasma PTH levels causing bone resorption, resulting in skeletal alterations. The susceptibility of different animal species to fibrous osteodystrophy varies, as does the distribution of lesions. Horses, pigs, dogs, cats, ferrets, dromedary camels, guinea pigs, reptiles, New World nonhuman primates, and goats are commonly affected, but the disease is rare in sheep and cattle1,4,5,6,9,12,13.
Primary hyperparathyroidism, typically caused by functional parathyroid gland adenoma, leads to an increase in PTH levels. Parathyroid gland adenocarcinoma and hyperplasia, although reported, are rare3,5. Generalized bone resorption can also occur because of paraneoplastic syndrome (pseudohyperparathyroidism or hypercalcemia of malignancy) when tumors produce calcitropic hormones that act similarly to parathyroid hormones2.
Secondary hyperparathyroidism is a much more frequent cause of fibrous osteodystrophy in animals than primary hyperparathyroidism and may stem from either chronic renal disease, or a dietary imbalance of calcium and phosphorus. PTH secretion is stimulated by a reduction in plasma ionized calcium, whatever the cause, and if the stimulus persists, generalized bone resorption results5.
Renal hyperparathyroidism occurs predominantly in small animals as a complication of chronic renal failure, most often in dogs and occasionally in cats. This alteration often referred as renal osteodystrophy, has recently been renamed “chronic renal failure-mineral and bone disorder” (CKD-MBD) in humans and veterinary medicine. Renal osteodystrophy may occur in dogs with CKD; however, the manifestations of uremia are usually more severe than those of skeletal lesions. Impaired glomerular filtration in renal failure leads to progressive hyperphosphatemia caused by reduced renal clearance of phosphate, causing hypocalcemia because of the inverse relationship between plasma ionized phosphate and calcium concentrations. Hyperphosphatemia also triggers the production of FGF23 by osteocytes, exacerbating hypocalcemia, as FGF23 functions to decrease the production of 1,25(OH)2D3 due to inhibition of the enzyme that acts in the formation and activates the enzyme that catabolizes 1,25(OH)2D3. Persistent hypocalcemia stimulates PTH release, resulting in osteoclastic bone resorption4,5,7.
Nutritional hyperparathyroidism while frequently noted in horses, is reported sporadically in other species, mainly in young, rapidly growing animals. This condition is generally caused by diet containing low calcium and a relatively high concentration of phosphorus, or in association with vitamin D deficiency. Vitamin D deficiency alone can cause rickets or osteomalacia, but reduced calcium absorption from the intestine in animals combined with vitamin D deficiency often results in concurrent fibrous osteodystrophy. Excess dietary phosphorus may cause fibrous osteodystrophy even in animals receiving adequate dietary calcium. Increased plasma phosphate concentrations, resulting from increased intestinal absorption of phosphorus, depresses plasma ionized calcium and indirectly stimulates the release of PTH. In all species, several factors influence the development and severity of lesions in secondary hyperparathyroidism. These include the degree to which dietary calcium is deficient and, perhaps more importantly, the degree to which dietary phosphorus is in excess. This condition usually occurs after ingesting unsupplemented diets consisting largely of grain, corn, and grain byproducts, such as bran, for some months, hence the term bran- disease1,5,8,9. In dogs and cats, nutritional hyperparathyroidism is often caused by diets that consist largely or entirely of meat or offal, as the calcium content of such diets is low, and the calcium to phosphorus ratio is very high. Additionally, in horses, fibrous osteodystrophy occurs in animals grazing on tropical grasses high in oxalate; even though dietary calcium and phosphorus are normal, oxalate binds calcium making it unavailable for absorption. Several grasses, including Setaria sphacelata, Cenchrus ciliaris, Brachiaria mutica, B. humidicola, Digitaria decumbens, Pennisetum clandestinum, P. purpureum, and Panicum spp., contain sufficient oxalate to produce clinical disease5,14.
The clinical signs and gross lesions of fibrous osteodystrophy generally develop more rapidly in young, growing animals because of their increased rates of bone synthesis and remodeling. Early signs include minor changes in gait, stiffness, transient and shifting lameness, and lassitude. Loss of appetite with progressive cachexia and decreased growth develops later. Affected animals frequently show bilateral enlargement of the bones of the skull, affecting both the maxillae and mandibles, hence the term big-head. Bony enlargement begins along the alveolar margins of the mandible, producing cylindrical thickening and reducing the intermandibular space. The molar margins of the maxillae subsequently begin to enlarge, and the enlargement spreads to involve the palate, remainder of the maxillae, and lacrimal and zygomatic bones. Initially, the enlargements were soft and could be cut using a knife. Involvement of the palate reduces the nasal passage and may cause dyspnea. Palatine and mandibular thickening causes a reduction in the buccal cavity, impairing mastication. This causes teeth to loosen and get partially buried or exfoliate, and the softened bone causes pressure, further impairing prehension and mastication1,5,9.
The microscopic features of fibrous osteodystrophy are similar in all domestic animal species, and characterized by increased osteoclastic bone resorption, marked fibroplasia, and increased osteoblastic activity with the formation of immature woven bone. In early lesions, the increase in bone resorption is reflected by an increased number of active osteoclasts, often within Howship’s lacunae along the surface of the trabeculae. Osteoblastic activity is also prominent, and it is not unusual to observe bone trabeculae resorption on one side, while new bone is being added to the other. Under physiological conditions, once osteoclasts have completed their required phase of resorption, they undergo apoptosis and disappear from the resorption sites. However, in fibrous osteodystrophy, groups of osteoclasts are frequently found mixed with fibroblastic elements as PTH enhances osteoclast survival1,5,9.
Contributing Institution:
Laboratório de Patologia Veterinária, Faculdade de Medicina Veterinária, Universidade Federal de Mato Grosso, Brazil.
JPC Diagnosis:
Jaw bone: Osteopenia, diffuse, severe, with marked fibroplasia (fibrous osteodystrophy).
JPC Comment:
The contributor provides an excellent review of the physiologic regulation of calcium and phosphorous and the pathologic manifestations of dysregulation of these minerals. Our scientific understanding of calcium and phosphorous metabolism began just over a century ago, but the descriptions of rickets date back millenia, with Roman physicians in the first and second century C.E. describing bowed legs and curved spines.11 Beginning in the 17th century, there was a steady increase in the number of children afflicted by rickets (or English disease, as it was known at the time). In 1634, rickets was listed as the cause of death in 14 of 10,900 deaths in London, and extensive texts describing the clinical and gross features of rickets were published around 1650.10,11 The association with lack of sunlight was first suspected by Polish physician Sniadecki in 1822, and in 1889, a map of cases in England revealed its association with industrialized cities, whose tall buildings and narrow streets blocked sunlight, and the seasonality of the affliction.8,10 In the 20th century, it was estimated that up to 90% of the urban-dwelling children in Boston and Leiden suffered from rickets.10,11
In the early 20th century, our understanding of calcium and phosphorous metabolism began to grow. The first histopathologic description of rachitic bones was published by German physician and pathologist Christian Georg Schmorl in 1909, and in 1918, British biochemist Edward Mellanby successfully induced rickets in beagle puppies by feeding an oatmeal diet.8,10 He further went on to cure the affliction by administering cod liver oil, a remedy used to preven trickets in fishing villages.8,10 Many suspected that the curative agent within cod liver oil was the fat soluble vitamin A. Elmer McCollum, who discovered vitamin B, hypothesized that a novel vitamin in cod liver oil possessed antirichitic properties; he proved this theory by demonstrating that cod liver oil still cured rickets even after aeration and heat destroyed the notoriously labile vitamin A.8,10,11 Around the same time, it was demonstrated that UV light exposure prevented and treated rickets; and exposure of certain foods to the same UV radiation endowed that food with antirichitic properties.8,10 Vitamin D2 and D3 were synthesized in the 1930s, and soon after, vitamin D supplementation of commercial foods and public awareness campaigns to increase sunlight exposure virtually eliminated rickets in industrialized nations.8,10 It was not until 2004 that FGF23 was discovered, which filled in many of the gaps in our knowledge of phosphorous metabolism.8
References:
- Bandarra PM, Pavarini SP, Santos AS, et al. Nutritional fibrous osteodystrophy in goats. Pesp Vet Bras. 2011;(31):10, 875-878.
- Bergman PJ. Paraneoplastic hypercalcemia. Top Companion Anim Med. 2012;27(4):156-158.
- Bonczynski J. Primary hyperparathyroidism in dogs and cats. Clin Tech Small Anim Pract. 2007;22(2):70-74.
- Chacar FC, Kogika MM, Zafalon RVA, et al. Vitamin D Metabolism and Its Role in Mineral and Bone Disorders in Chronic Kidney Disease in Humans, Dogs and Cats. Metabolites. 2020;10(12):499.
- Craig, LE, Dittmer, KE, Thompson, KG. Bones and joints. In: Maxie, MG, ed. Jubb, Kennedy, and Palmer’s Pathology of Domestic Animals. 6th ed. St Louis, MO: Elsevier; 2016:16-163
- Dittmer KE, Thompson KG. Vitamin D metabolism and rickets in domestic animals: a review. Vet Pathol. 2011;48(2):389-407.
- Foster JD. Update on Mineral and Bone Disorders in Chronic Kidney Disease. Vet Clin North Am Small Anim Pract. 2016;46(6):1131-1149.
- Gallagher JC, Rosen CJ. Vitamin D: 100 years of discoveries, yet controversy continues. Lancet Diabetes Endocrinol. 2023; 11(5): 362-374.
- Hines ES, Stevenson VB, Patton ME, et al. Fibrous osteodystrophy in a dromedary camel. J Vet Diagn Invest. 2021;33(1):144-148.
- Holick MF. The One-Hundred-Year Anniversary of the Discovery of the Sunshin Vitamin D3: Historical, Personal Experience, and Evidence-Based Perspectives. Nutrients. 2023; 15(3): 593.
- Miller WL, Imel EA. Rickets, Vitamin D, and Ca/P Metabolism. Horm Res Paediatri. 2022; 95: 579-592.
- Schwarz T, Störk CK, Megahy IW, et al. Osteodystrophia fibrosa in two guinea pigs. J Am Vet Med Assoc. 2001;219(1):63-66.
- Tokarnia CH, Döbereiner J, Peixoto PV. Poisonous plants affecting livestock in Brazil. Toxicon. 2002;40(12):1635-1660.
- Uhl EW. The pathology of vitamin D deficiency in domesticated animals: An evolutionary and comparative overview. Int J Paleopathol. 2018;23:100-109.